- 2.1 Introduction
- 2.2 Different Forms of DNA Polymorphisms
- 2.3 Human Evolution with Special Reference to Mitochondrial DNA and Y-Chromosome Polymorphisms
- 2.3.1 mt DNA Polymorphism-Human Evolution
- 2.3.2 Y Chromosome Polymorphism-Human Evolution
- 2.4 DNA Polymorphisms and Disease Association
- 2.4.1 Monogenetic Disease
- 2.4.2 Multifactorial Disease
- 2.5 Techniques in Molecular Genetics
- 2.5.1 Polymerase Chain Reaction
- 2.5.2 Restriction Fragment Length Polymorphism
- 2.5.3 DNA Sequencing Methods
- 2.5.4 Microarray
2.1 INTRODUCTION
Human genome (entire genetic material present in a cell) consists of 3 billion bases. There are about 10 million Single Nucleotide Polymorphisms. The DNA sequence will carry code of information for carrying genetic information from parent to child (generation to generation). A person or plant or animal phenotypically looking different, means that there are variations in the genetic material of the organism. Any change in the DNA sequence will bring change in the genetic information, inturn brings change in phenotypic expression and biological function. The change in the DNA sequence is called Mutation. If the mutation frequency is more than 2 per cent in a population, it is called as polymorphism. Hence, we can define DNA polymorphism as DNA having more than one form, with a frequency of above 2 percent in a population.
2.2 DIFFERENT FORMS OF DNA POLYMORPHISMS
DNA polymorphisms can be studied in the form of Single Nucleotide Polymorphisms (SNPs), Restriction site Polymorphisms (RSPs) or Restricted Fragment Length Polymorphisms (RFLP) and Variable Number of Tandem Repeats (VNTRs).
Single Nucleotide Polymorphisms (SNPs): A single nucleotide is substituted by a different nucleotide.
There are two types of nucleotide substitutions resulting SNPs. 1) Transition: A substitution occurs between Purines (A,G) or between Pyrimidines (C,T). This type of substitution constitutes two thirds of all SNPs. 2) Transvertion: A substitution occurs between a Purine and a Pyrimidine.
Insertions: A new nucleotide will be inserted in the sequence.
Deletions: An existing nucleotide will be deleted in the sequence.
Restriction site Polymorphisms (RSPs) or Restricted Fragment Length Polymorphisms (RFLP): A sub set of SNPs cause a loss or gain of a restriction site (restriction site is the location where a particular enzyme cuts the DNA sequence at particular sequence location into pieces of DNA). Due to change in nucleotide at particular site location, enables the enzyme to cut DNA into pieces. This leads to create different length of DNA piece in an individual and another length of DNA piece in another individual. This is called Restriction site polymorphism. RSPs are described as restricted fragment length polymorphisms (RFLP).
III.Variable Number of Tandem Repeats (VNTRs): It is divided into two types:
Microsatellite polymorphism
Minisatellite polymorphism.
Micro-satellite polymorphism is also called as Short Tandem Repeats (STRs). A small array of tandem repeats of a simple sequence (usually less than 10 base pairs). Ex; GATAA GATAA GATAA GATAA GATAA GATAA in this sample 5 bases repeated 6 times.
a) GA GA GA GA GA – it is dinuleotide repeat
b) TAT TAT TAT TAT- it is trinuleotide repeat
Mini-satellite polymorphism: A collection of moderately sized arrays of tandemly repeated DNA sequence which are dispersed over considerable portions of the nuclear genome.
c) TTAGGGTACCGG TTAGGGTACCGG TTAGGGTACCGG –this array of 12 nucleotides repeats from 3-20 Kbp(thousand base pairs).
2.3 HUMAN EVOLUTION WITH SPECIAL REFERENCE TO MITOCHONDRIAL DNA AND Y-CHROMOSOME POLYMORPHISMS
DNA polymorphisms particularly SNPs became a powerful tool in reconstructing human origins, evolution and prehistoric migrations. Earlier we used to depend on archaeological and paleontological evidences to reconstruct human evolution. In the absence of these traditional evidences DNA analysis became an alternative tool to reconstruct human evolution.
The hominid fossil record in Africa begins about 4 MYs ago in Early Pliocene with representatives of the genus Australopithecus from Ethiopia and Tanzania. Homo erectus arose more than a million years ago in the Pleistocene, giving rise to our own genus, Homo. Anatomically modern humans began to appear 120000100000 years ago and co-existed with Neanderthals until the latter became extinct about 30,000years ago. Based on these evidences, two theories have been proposed for the evolution of modern humans: 1.Multiregional evolution: It proposes that present day worldwide populations are the descendants’ of in situ evolution after an initial dispersal of Homo erectus from Africa during the Lower Pleistocene(~650kybp), 2. Uni regional hypothesis (also called as Recent African Origin model or Out-of-Africa):- All present day populations have descended from a recent common ancestor that lived in East Africa ~150,000 years ago.
At this juncture, mitochondrial DNA and Non-recombining region of Ychromosome (NRY) analysis provided an alternative approach to reconstruct Modern Human evolution. mtDNA and NRY Y-chromosome analysis enable us to trace maternal and paternal lineages of modern humans. Along with these DNA markers autosomal and X-linked markers have also been studied.
DNA can also be extracted from bone material of ancient specimens. The ancient mtDNA analysis from Neanderthal specimens reveals that Neanderthals are not immediate ancestors to modern humans. Modern humans diverged from Neanderthals about 400,000 years ago. Neanderthals went extinct without contributing any mtDNA to modern humans.
2.3.1 mtDNA Polymorphism-Human Evolution
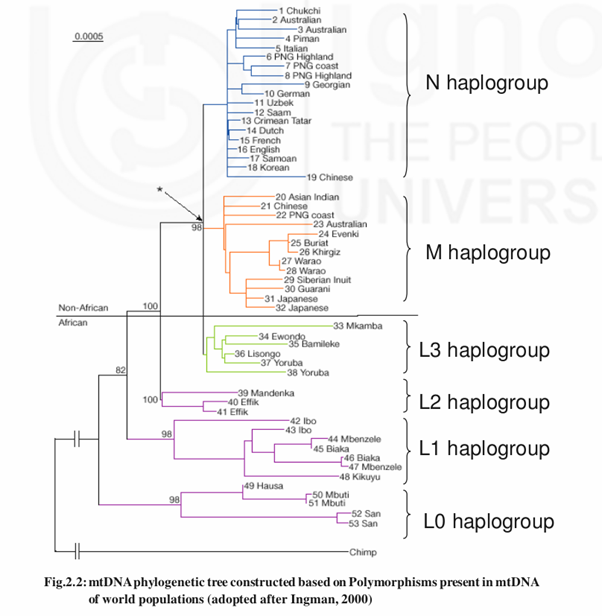
DNA polymorphisms suggested a recent origin of modern humans from African populations. Initial evidence came from mtDNA, which is transmited maternally. Each human cell cytoplasm contains 10-100 mitochondria. Each mitochondrion will have a circular double strand DNA molecule about 16569 base pair length. The most ancient mtDNA haplotypes (having the same genotype) are L0, L1, L2
and L3. Haplotypes L1 and L2 are specific to the sub-Saharan Africa. L3 is present in North East Africa and Middle East. L3, M & N are parallel branches. M branch is called as Asian branch and N is called as European branch. All branches of M arose in Asia. M branches didn’t present in Europe. M1, a branch of M present in East Africa is originated in Middle East and back migrated into Africa. In Asia N branch is also present. M is the oldest branch than N branch. An ancestral branch of Asian might have arisen in North East Africa and subsequently left to colonise Asia (50-70 thousand years ago) and Europe (4550 thousand years ago).The mtDNA analysis of world populations reveals that modern humans can be traced back to a single mother, ‘mitochondrial eve’. The analysis also shows that this individual existed about 100,000-130,000 years ago in east Africa. Of course, the mitochondrial eve was not the only person living on the planet at that time: there are perhaps about 10,000 individuals living at that time but unlike Eve, there mtDNA sequences didn’t get transmitted to the present human populations.
SNPs normally exhibit two forms of variation at a nucleotide position or have two alleles. For example Africans (L0, L1 & L2 haplogroups) have Thymine at nucleotide position 3594 where as all Non-Africans will have Cytocine at np3594. Likewise all Asians have four mutations (M lineage) in their mtDNA sequence
Indian scenario
The initial dispersal of modern humans from East Africa en-route North and East of Africa has now been documented, following the African mtDNA haplogroups into Saudi Arabia and then Western India. Indian specific mtDNA branches, M and N encompass all the populations in India irrespective of their social rank, caste or tribe. In India the frequency of M haplogroup ranges from 54 to 97 percent. Indian specific M sub haplogroups are M2,-M6, M18, M25, and M30-M62. M1is present in North East Africa, M31 & M32 are specific for Andaman Islands. M7, M8, M9, M10, M11 & M12 are specific for China and Japan. Most numerous sub haplogroups of European N haplogroup are Indian specific. Ex: N5, R5, R6, R7, R8 T30, R31, U2. Genetic links of Indians with East Eurasians, West Eurasians and Australians are established by mtDNA polymorphisms.
2.3.2 Y Chromosome Polymorphism-Human Evolution
The Y chromosome is a suitable tool for investigating the recent human evolution, for medical genetics, DNA forensics and genealogical reconstructions, due to its uniqueness among the other human chromosomes. The Y chromosome has a sex-determining role, it is male specific and constitutively haploid (Single). It is inherited paternally and is transmitted from father to son, and unlike other chromosomes, the Y chromosome escapes meiotic recombination in its NRY (Non Recombining Y chromosome) region. The non-recombining portion of the Y chromosome descends as a single locus. As they change only by accumulating mutations in time, they preserve by far more simple record of their history compared to autosomes.
Y chromosome variation consists of large amount of different types of polymorphisms, which are widely used in evolutionary studies. They may roughly be divided into two large groups: bi-allelic markers and polymorphisms of tandem repeats or multi-allelic markers. Bi-allelic markers include SNPs (Single Nucleotide Polymorphisms) and insertions and deletions (indels). SNPs are the most common type of polymorphisms, constituting more than 90% of total polymorphisms of DNA. Only these bi-allelic mutations that have occurred, only once in history of humans and have a detectable frequency in human populations are used in phylogenetic studies.
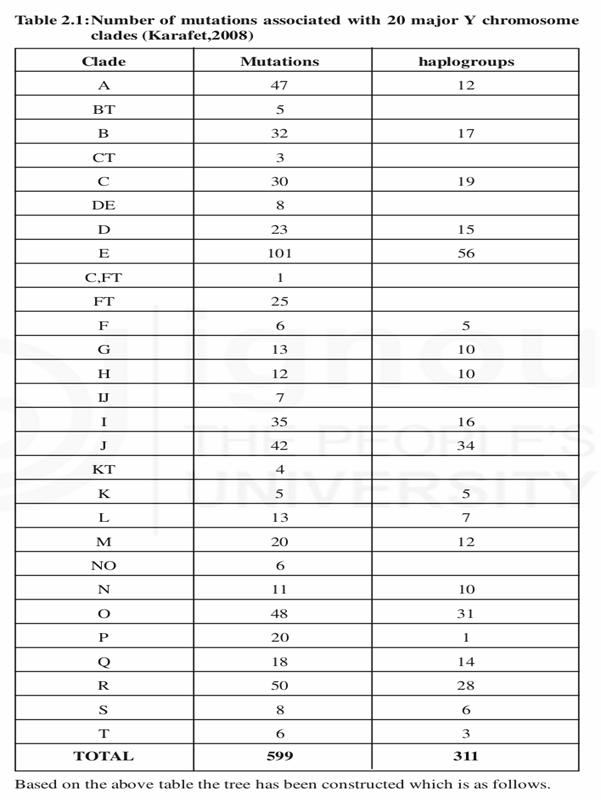
Y chromosome DNA polymorphisms are useful to trace paternal lineages. The Y chromosome consortium is formed to document the binary polymorphisms in NRY (non recombining region of Y chromosome). There are about 599 polymorphisms made Y chromosomes in to 311 groups. This information was constructed into a tree and named the main branches starting from alphabets A to T. Major branches are called as Clades and sub branches are called as a haplogroups.
Polymorphisms at P91, M168, M294 distinguishes A, B clades from the rest of clades. Clade A & B are exclusively present among African populations. The majority of branches of the Y chromosome tree outside Africa are composed of a tripartite assemblage of the following haplogroups: a) C; b) D and E, and c) an overarching haplogroup F that defines the internal node of all remaining haplogroups from G to T.
Because the mutation defining haplogroup C (M130=RPS4Y) has not been observed in any African populations, this haplogroup is likely to have arisen somewhere in Asia after an early departure of modern humans from Africa, prior to the arrival of them to Sahul in Southeast Asia. The most western region where haplogroup C* has been detected is India. This lineage consists of several sub lineages with irregular phylogeographic patterning, ranging from Central and North Asia to America and in the direction of Southeast Asia up to Australia and Oceania. Differently from hg C, haplogroups E and D share three phylogenetic ally equivalent markers.
Calde D* is found in Andaman Islands whereas D1 & D2 are found in Tibetans and Japanese. E is the most frequent and divergent in Africa.The third major sub-clade of M168 lineages is super haplogroup F. It is characterized by mutation M89 at its root from which all other haplogroups deploy. F has been suggested to have evolved early in the diversification and migration of modern humans. Later on, the ancestral trunk of F diversified into many branches by subsequent acquisition of mutations, giving rise to many region-specific haplogroups, such as J and G in Near and Middle East, I in Europe, H in Southern Asia, etc. An expansion of F lineages gave rise also to a population that acquired the M9 mutation (haplogroup K), which defines another major bifurcation in the phylogeny . The branches of this clade probably migrated in different directions (North and East) and gave start to many separate and region-specific haplogroups in Eurasian continent and beyond. Out of descendants of M9 lineage, haplogroup L (M20) has greatest frequency in Southwest Asia and distinctive K lineages and M (M4, M5) haplogroup are restricted to Oceania and New Guinea, whereas haplogroup O with its numerous sub-clades predominates in southern and southeastern Asia, reaching North China, Manchuria and some Siberian populations. The population carrying M9 expanded also in direction of north towards Central Asia characterized by subsequent mutations defining haplogroup P, which encompasses distinctive eastward expanding haplogroup Q (M242) characteristic to Siberian populations and Amerindians and Eurasian haplogroup R lineages that have expanded westward. Thus, one may speculate that multiple independent formations and fragmentations of populations carrying F-related lineages throughout most of Eurasia may have displaced the earlier haplogroup C and D lineages towards the margin in many areas. Among Indians H, O, R1, R2 are the major clades. H haplogroup is nearly restricted to India, Srilanka and Pakistan. Among Austro-Asiatic language speaking groups of India O haplogroup is predominant followed by H group. Indo-European language speakers have 50 percent of O haplogroup followed by H & R.
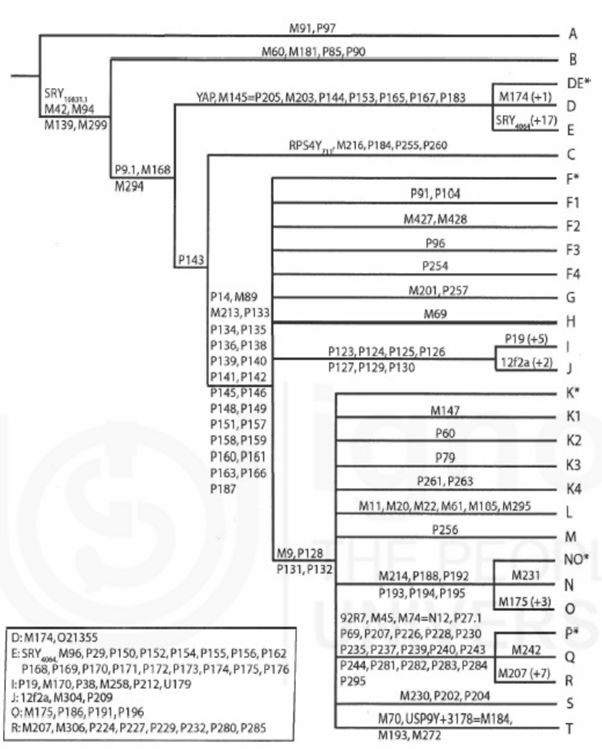
Apart from bi-allelic polymorphisms, insertions and deletions (indels) persist over generations and are sufficiently common to be considered as polymorphisms. One such example is a 2kb deletion in 12f2 marker, used for defining haplogroup J. Some indels have arisen independently more than once in human history. For example, the deletion or duplication of the 50f2/C region in background of different haplogroups is thought to be arisen at least 7–8 times. Another example is the deletion of DAZ3/DAZ4 region that has been indicated to occur in haplogroup N individuals, widely spread in northern Eurasia.
Another frequent type of polymorphism, present also in Y chromosome, is tandem repeats, mostly in non-coding DNA regions. According to their length, these repeats are classified as satellite-DNAs (repeat lengths of one to several thousand base pairs), mini-satellites or variable number of tandem repeats (VNTRs), ranging from 10 to 100 bp, and microsatellites or short tandem repeats (STRs), with motifs less than 10 bp, mostly 2 to 6 bp long. In Y-chromosomal studies microsatellites are widely used, than mini-satellites. Microsatellites are multiallelic markers with different allele numbers ranging from 3 to 49 in locus. Their mutation rate is much higher than that for biallelic markers and, therefore, they are widely used in phylogenetic studies to investigate details of demographic events that have occurred in a more recent time scale. In evolutionary studies STRs are valuable in combination with binary haplogroup data, as they enable us to study diversity within a haplogroup. STRs are particularly widely explored in forensic work. So far the number of widely used Y chromosomal STRs has been quite low (about 30) but in a recent study 166 new and potentially useful STRs were described.
Based on the Phylogenetic analysis it was concluded that, all humans have originated from an African ancestor. About 70,000 thousand years ago modern man came out of Africa and peopled all the continents.
Thus the combination of molecular age and geographical structure makes mtDNA and the NRY a sensitive genetic index capable of tracing the micro evolutionary patterns of noval modren human diversity. Mitochondrial DNA and Y chromosome studies in Indian populations reveals affiliation with Europeans, East Asians, Austro-Melanesians and in situ development of deep rooted ancestry whose relative clustering and coalescence ages suggest shaping of Indian gene pool during late pleistocene.
2.4 DNA POLYMORPHISMS AND DISEASE ASSOCIATION
Disease is a disordered or incorrectly functioning of organ, part, structure, or system of the body resulting from the effect of genetic or developmental errors, infection, poisons, nutritional deficiency or imbalance, toxicity, or unfavourable environmental factors.
A genetic disease is any disease that is caused by an abnormality in an individual’s genome. The abnormality can range from minuscule to major from a discrete mutation in a single base in the DNA of a single gene to a gross chromosome abnormality involving the addition or subtraction of an entire chromosome or set of chromosomes. Some genetic disorders are inherited from the parents, while other genetic diseases are caused by acquired changes or mutations in a preexisting gene or group of genes. Mutations occur either randomly or due to some environmental exposure. There are about 30,000 genes in humans. Some of the mutations in these genes cause a disease, predispose us to the common diseases in combination with other variants and with the environment. Knowledge of these polymorphisms offers tremendous advantage in the study of disease and variable response to treatment.
It is caused by changes or mutations that occur in the DNA sequence of a single gene, also called Mendelian disorder. There are more than 6,000 known singlegene disorders, which occur in about 1 out of every 200 births. Some examples of monogenetic disorders include: Cystic Fibrosis, Sickle Cell Anaemia, Marfan syndrome, Huntington’s disease, and Hemochromatosis. Single-gene disorders are inherited in recognisable patterns: autosomal dominant, autosomal recessive, and X-linked.
Example: Sickle cell anaemia is a disease passed down through families in which red blood cells form an abnormal crescent shape (Red blood cells are normally shaped like a disc.) Sickle cell anaemia is caused by an abnormal type of haemoglobin called haemoglobin S. Haemoglobin is a protein inside red blood cells that carries oxygen. Haemoglobin S changes the shape of red blood cells, especially when the cells are exposed to low oxygen levels. Then the red blood cells become crescent shaped or sickles. The sickling occurs because of a mutation in the haemoglobin gene. The haemoglobin beta(HBB) gene is found in region 15.5 on the short (p) arm of human chromosome 11. In sickle cell haemoglobin (HbS) the glutamic acid in position 6 is mutated to valine in a beta chain. This change allows the deoxygenated form of the haemoglobin to stick to itself and become crescent shape.
The fragile, sickle shaped cells deliver less oxygen to the body’s tissues. They can also get stuck more easily in small blood vessels, and break into pieces that interrupt healthy blood flow.
Sickle cell anaemia is inherited from both parents. If you inherit the haemoglobin S gene from one parent and normal haemoglobin (A) from your other parent, you will have sickle cell trait. People with sickle cell trait do not have the symptoms of sickle cell anaemia. The children of both sickle cell parents will get sickle cell anaemia.
Sickle cell disease is much more common in people of African and Mediterranean descent. It is also seen in people from South and Central America, the Caribbean, and the Middle East.
2.4.2 Multifactorial Disease
It is called complex or polygenic disease. Complex diseases are caused by a interaction of environmental factors and mutations in multiple genes. Some common chronic diseases are multifactorial in nature. Examples of complex diseases include: Cardio Vascular diseases, high blood pressure, Alzheimer’s disease, arthritis, diabetes, cancer, and obesity. For example, different genes that influence breast cancer susceptibility have been found on chromosomes 6, 11, 13, 14, 15, 17, and 22.
Mutations in BRCA1 gene (BRCA1 Gene is located on chromosome17q21.31) contribute significantly to the development of familial/hereditary breast and ovarian cancer. Founder mutations such as the BRCA1-185delAG and 5382insC are found among Ashkenazi Jews.
Polymorphism in BRCA1 Chr17 at np 37043496 is shown in the figure.
CCGCCCCTACCCCCCGTCAAAGAATACCCAT(normal form)
CCGCCCCTACCCCCCCTCAAAGAATACCCAT (mutated form)
Large rearrangements, mostly deletions in regions of Y-specific genes (AZFa, AZFb, AZFc), have been known as causes for many diseases leading to male infertility, causing spermatogenic failure, azoospermia, severe oligo spermia or otherwise severely impair male reproductive fitness.
2.5 TECHNIQUES IN MOLECULAR GENETICS
To study DNA polymorphisms initial step is extraction of DNA from cells. We can extract up to 1500 nano grams of DNA from 5ml of blood by Phenol chloroform method.
DNA Extraction- Principle
The process of DNA extraction can be divided into three stages: (i) disruption of the cellular membranes, resulting in cell lysis, (ii) protein denaturation, and finally (iii) the separation of DNA from the denatured protein and other cellular components.
About 5 ml of blood is transferred to a sterile 15ml conical bottom polypropylene tube and equal amount of RBC lysis buffer is added. Shake the tube gently for 3 to 5 minutes. The contents of the tube transforms into transparent red colour upon the lysis of RBC. We have to ensure that RBC is completely lysed before proceeding to further step. Once RBC is lysed, transfer the tubes into the centrifuge. Run the centrifuge at 1500 rpm for 15 mts at 20oC. Upon the completion of the run carefully, remove the tubes from the centrifuge and decant the supernatant without disturbing the pellet at the bottom. Now add 4 ml of RBC buffer to the tube and break the pellet using hand or vortex. Repeat the centrifugation step as earlier with same settings. Decant the supernatant, if pellet is still red repeat the RBC lysis. If pellets are light pink or white proceed for further step.
Add 1 ml of digestion buffer and 10 ul of Proteinase K to the tube and carefully dislodge the pellet from the bottom of the tube. Adjust the hot water bath at 55oC, now transfer the tubes into the hot water bath. Gently dislodge the tubes for every 30 minutes to enhance the digestion process. The content of the tubes turns clear and transparent upon the digestion.
Now add 250ul of 5M sodium per chlorate to the tube and gently mix the contents by partially inverting the tube. Now add 500ul of tris saturated phenol, 500ul of 24:1 chloroform isoamyl alcohol, mix the contents thoroughly and adjust the centrifuge at 4ºC, 4000 rpm and 15 minutes. Take a fresh tube, carefully transfer the supernatant using the 1ml pipette and cut tips into the fresh tube. Now add 500ul of 24:1 chloroform and isoamyl alcohol, and repeat the centrifugation step with same settings. Carefully transfer the supernatant using a cut tip into a fresh tube. Add double the volume of chilled alcohol; gently invert the tube for a minute. A milky white fibrous DNA is visible. Now transfer the DNA into a 1.5ml tube, add 1 ml of 70 % alcohol, spin at 12,000 rpm for 10 minutes, and repeat the step for one more time to eliminate the remaining protein contamination. Dry the pellets and add 200ul of TE buffer and mix the contents thoroughly and transfer into the hot water bath/dry bath for digesting the DNA. This process usually takes 2 hours. Transfer tubes for -80ºC for long term storage.
Details of the buffers and reagents used for DNA extraction are given below.
1) RBC Lysis Buffer contains Sucrose, 1M Magnesium Chloride 1M Tris-Hcl and Triton X.
2) Digestion Buffer constitutes 1M Tris-Hcl (pH 8.0), 1M Sodium Chloride, 0.5M EDTA (Na salt)
2) Tris-EDTA Buffer made up of 1M Tris-HCl (pH 8.0), 0.5M EDTA
After DNA was completely dissolved in the TE buffer, its quantity and quality was checked by both spectrophotometry and gel electrophoresis.
Determination of DNA concentration by Spectrophotometry
Prior to any analysis, DNA should be quantified and checked for purity and integrity. Based on its structure, DNA absorbs light in the ultraviolet range,
specifically at a wavelength of 260nm. A value of 1 at OD260 is equal to 50ng/µl double stranded DNA. Therefore to calculate the concentration of DNA, the following formula can be used:
Concentration of DNA = 260nmabs x 50ng/µl
Procedure
2µl DNA sample was diluted to 200 µl with Double Distilled water (Dilution 1:100). Spectrophotometer was set to auto zero with the Double Distilled water. Optical Density (OD) of the diluted DNA aliquot was measured at 260 nm and 280nm using quartz crystal cuvette.
Quality Assessment
A ratio of OD values at 260nm and 280nm indicates the purity of the extracted DNA sample. If the ratio is within range of 1.6 to 2.0, then DNA sample is considered as clear and free from contaminants like residual protein and mRNA. An OD ratio less than 1.6 indicate the residual proteins or phenol contamination, whereas ratio of more than 2.0 indicates residual RNA contamination.
Quantity Assessment
DNA quantity was estimated as the OD value at 260nm of extracted sample is 1.00 then the concentration of the DNA is 50ug/ml.
Therefore, DNA concentration = OD at 260nm x 50 x Dilution factor.
DNA quantification and electrophoresis
Electrophoretic analysis of DNA using agarose gels can confirm DNA integrity. Typically intact genomic DNA will be up to 40KB in size, depending upon the species. Prepare 1% agarose gel has to be by adding required quantity of agarose to 1X Tris-Acetate-EDTA (TAE) buffer and mix well. Heat the mixture in microwave oven until it became clear and take care to avoid over boiling and evaporation. Cool the mixture to ~500 C and add ethidium bromide to make a final concentration of 0.001ug/ml. Pour the entire mixture into a tray in which combs are fixed to make wells in the gel. After gel formation, place the tray in buffer tank containing 1X TAE buffer for submerged gel electrophoresis and remove the combs with care to avoid rupture of wells. Mix 1µl of each DNA sample with 1µl of loading dye and load the mixture into the wells. Subject the Gel to electrophoresis at 90V for 30 minutes and visualise using gel documentation system where it is exposed to Ultraviolet rays. Under Ultraviolet rays exposure, DNA will give luminance which indicate the presence of DNA in the sample as shown in the below figure.
2.5.1 Polymerase Chain Reaction
PCR (Polymerase Chain Reaction) is a revolutionary method developed by Kary Mullis in 1980s. It is an essential and ubiquitous tool in genetics and molecular biology. With the use of this technique we can clone DNA invitro.
PCR is based on using the ability of DNA polymerase to synthesize new strand of DNA complementary to the offered template strand. Because DNA polymerase can add a nucleotide only onto a pre existing 3′-OH group, it needs a primer to which it can add the first nucleotide. This requirement makes it possible to delineate a specific region of template sequence that the researcher wants to amplify. At the end of the PCR reaction, the specific sequence will be accumulated in billions of copies (amplicons). DNA sequencing by dye termination technique requires multiple copies of DNA, hence PCR is performed to generate numerous copies of DNA fragments of interest which were further used for sequencing.
The genomic DNA of each subject can be amplified on thermal cycler with an initial denaturation at 960C for 3 minutes and later on for 35 cycles at 950C for 60 seconds, at estimated annealing temperature of the primer for 45 seconds, extension at 720 C for 2.30 minutes and a final extension at the end of 35th cycle at 720C for 7 minutes in a final volume of 10 µl containing 50mM KCl, 10mm Tris (pH 8.3), 1.5mM MgCl2, 75 ng of each primer, 100 µM deoxy-NTP, and 1 U Taq polymerase.
Primer designing
The main limitation of PCR technique is to provide short pieces of single-stranded DNA (primers) that are complementary to a part of target sequence. With the use of human genome sequence available we can now design primers to any region of interest in the human genome. The most critical step in PCR experiment is designing oligo-nucleotide primers. As poor primers could result in little or even no PCR product, alternatively they could amplify unwanted DNA fragments. Either will affect the downstream analysis. Many of the factors which affect the primers specificity and sensitivity like product size, primer size, tm, GC content, GC clamps and dimer formation can be adjusted as per the user requirement. Primers which fit the specified criteria can be checked for their specificity using NCBI BLAST.

2.5.2 Restriction Fragment Length Polymorphism
Restriction-Fragment Length Polymorphism (RFLP) was proposed by American geneticist David Botstein, biochemist Ronald W. Davis, population geneticist Mark Skolnick, and biologist Ray White. Restriction fragment length polymorphisms (RFLPs) can be used to produce a linkage map of the human genome and to map the genes that cause disease in humans.
Restriction Fragment Length Polymorphism (RFLP) analysis measures fragments of DNA containing short sequences that vary from person to person, called VNTRs. After extracting DNA from a sample and amplifying it with the technique known as Polymerase Chain Reaction, we can add restriction enzymes that cut the DNA at specific points. The resulting fragments can be sorted by length with gel electrophoresis technology to determine how many times a given VNTR is repeated.
If two different samples show VNTRs of different lengths, the samples could not have come from the same person. On the other hand, two samples showing VNTRs of the same length could have come from the same person, or from two people who happen to have VNTRs of the same length at that location. By comparing enough VNTRs from two individuals, however, the likelihood of a coincidental match can be reduced to nearly zero. RFLP testing requires hundreds of steps and weeks to complete, and it has been largely replaced by newer, faster techniques.
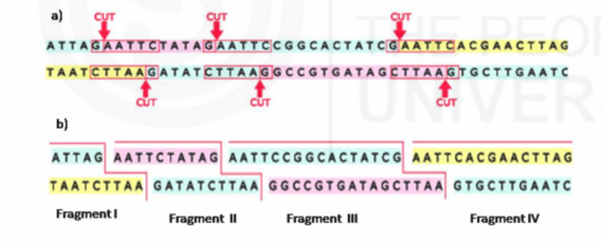
Short Tandem Repeats in DNA Analysis
STRs can be amplified and sequenced using PCR and Sequencing techniques. Analysis is based on the number of repeats present in the sample.
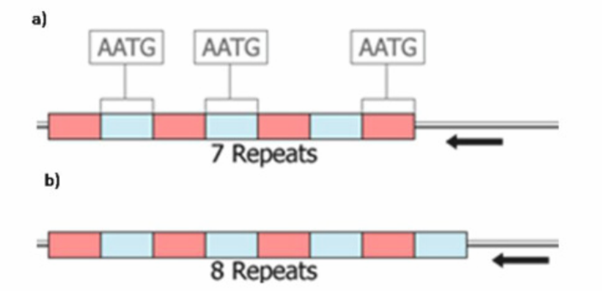
2.5.3 DNA Sequencing Methods
In the 1960s and 1970s, British scientists Frederick Sanger and Alan Coulson, Alan Maxam and Walter Gilbert in the United States, developed DNA sequencing techniques. Automated equipment makes DNA sequencing a speedy, routine laboratory procedure. Sanger and Gilbert won the 1980 Nobel Prize in Chemistry for their work.
Sanger method of DNA Sequencing
In Sanger method, specific terminators of DNA chain elongation 2’, 3’dideoxynucleoside triphosphates are synthesized. These molecules can be incorporated normally into a growing DNA chain through their 5’- triphosphates groups. However, they cannot form phosphodiester bonds with the next incoming deoxynucleotide triphosphates (dNTPs). When a small amount of a specific dideoxy NTP is included along with the four deoxyNTPs normally required in the reaction mixture for DNA synthesis by DNA polymerase, the products are a series of chains that are specifically terminated at dideoxy residue. This forms the basis for Sanger’s method.
Procedure
Initially single strand DNA is prepared through denaturation process. Then single strand DNA is mixed with a short end labeled piece of DNA (Primer) that is complementary to the end of single strand DNA. Labeling of primer is carried out using enzymes like Alkaline Phosphatase and Polynucleotide Kinase. After primer is annealed to DNA, sample is divided into four portions in four tubes. In each tube, along with DNA, Primer, DNA polymerase, a carefully controlled ratio of one particular dideoxynucleotide with its normal deoxynucleotide, and the other three dNTPs are added.
In each tube, DNA polymerase polymerizes normally from primer by utilizing nucleotides. When ddNTP is incorporated, the growth of that chain will stop. If the correct ratio of ddNTP: dNTP is chosen, a series of labeled strands will result, the lengths of which are dependent on the location of a particular base relative to the end of the DNA.
After suitable time period, the resultant labeled fragments in each tube are separated by size on an acrylamide gel. The separated fragments are detected by exposure of the gel to x-ray film through the process of autoradiography. Fromthe band developed in each lane of the autoradiograph and knowledge of which lane contain which base, the sequence of the complementary sequence can be obtained. From the complementary sequence, the sequence of the original strand can be easily determined with the help of Watson and Crick base pairing rule. Thus Sanger method is used for DNA sequencing.
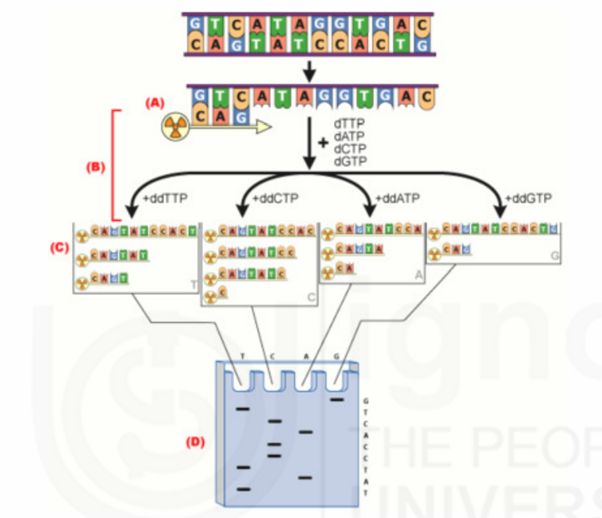
Automated DNA Sequencing
There are various methods available for DNA sequencing like chemical degradation, chain termination method, sequencing by ligation and micro fluidic Sanger sequencing etc. Advances in automation have opened gates to new fast and reliable automated DNA sequencing technologies. Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Dye-terminator sequencing is a slight modification of the Sanger’s chain termination method it utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, with different wavelengths of fluorescence and emission. The dye labelled DNA fragments will be capillary electrophoresed and a detection system will identify the labelled bases when they pass through a laser that activates the dye.
Cycle sequencing
To read the sequence of the amplified DNA termination with florescent dye labelled ddNTPs, each base PCR amplicon should be subjected to cycle reaction with one primer using BigDye terminator cycle sequencing ready reaction kits following the manufacturer’s guidelines.
Sequencing run
The PCR product should be added 10µl of Hi-Di formamide before feeding it to the sequencer machine.
Sequence Alignment
The generated sequences can aligned to their respective reference sequences with the use of software called DNA baser. It performs sequence comparisons for variant identifications, SNP discovery and validation. It allows analysis of the re-sequenced data, comparing the consensus sequences to a known reference sequence. The reference sequences for the gene studied are obtained from NCBI Gen bank data base.
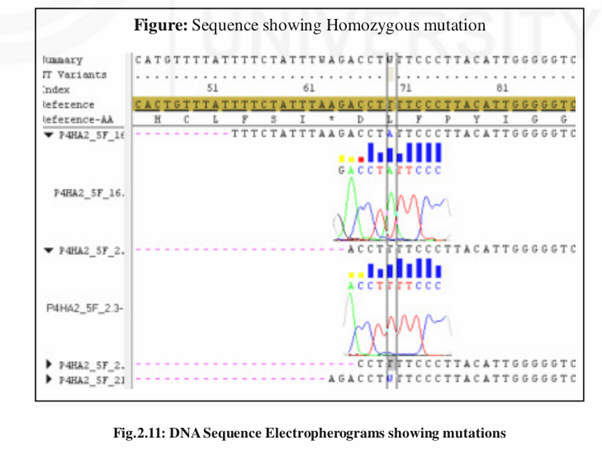
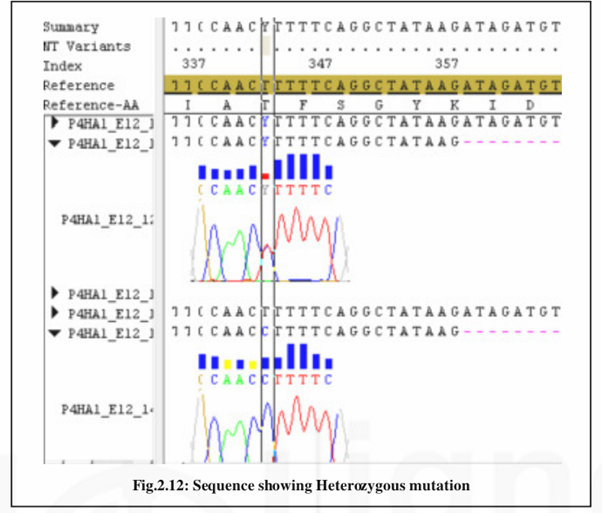
Sequence Editing and Mutation Scoring
We can score the mutations from the aligned sequences by checking electropherograms of the DNA sequences. Genotypes can be exported from the software for further analysis.
Single Nucleotide Polymorphisms or SNPs (pronounced snips”) are variations in a DNA sequence that occur when a single nucleotide in the sequence is different from the normal in at least one percent of the population. When SNPs occur inside a gene, they create different variants or alleles, of that gene.
Unlike repeated portions of DNA like STRs and VNTRs, in the case of SNPs it is the sequence itself, not its length that is useful to forensic scientists. SNPs are commonly occurring every 100 to 300 bases along the entire length of the human genome. Mutations in SNPs are very rare, so the sequences tend to be passed unchanged across generations. But because any given SNP is relatively common in the population, an analyst must examine dozens of SNPs to derive a true DNA fingerprint. For this reason, SNP analysis is rarely used in forensic cases.

2.5.4 Microarray
A DNA microarray (also commonly known as gene chip, DNA chip, or biochip) is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles (10-12 moles) of a specific DNA sequence, knownas probes (or reporters). These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA sample (called target) under high-stringency conditions. Probe-target hybridisation is usually detected and quantified by detection of fluorophore, silver, or chemiluminescence-labelled targets to determine relative abundance of nucleic acid sequences in the target
DNA microarrays can be used to measure changes in expression levels, to detect single nucleotide polymorphisms (SNPs), or to genotype or resequencemutant genomes (see uses and types section).
Principle
Hybridisation of the target to the probe
The core principle behind microarrays is hybridisation between two DNA strands, the property of complementary nucleic acid sequences to specifically pair with each other by forming hydrogen bonds between complementary nucleotide base pairs. A high number of complementary base pairs in a nucleotide sequence means tighter non-covalent bonding between the two strands. After washing off of non-specific bonding sequences, only strongly paired strands will remain hybridized. So fluorescently labelled target sequences that bind to a probe sequence generate a signal that depends on the strength of the hybridisation determined by the number of paired bases, the hybridisation conditions (such as temperature), and washing after hybridisation. Total strength of the signal, from a spot (feature), depends upon the amount of target sample binding to the probes present on that spot. Microarrays use relative quantisation in which the intensity of a feature is compared to the intensity of the same feature under a different condition, and the identity of the feature is known by its position.
Many types of arrays exist and the broadest distinction is whether they are spatially arranged on a surface or on coded beads: (i) The traditional solid-phase array is a collection of orderly microscopic “spots”, called features, each with a specific probe attached to a solid surface, such as glass, plastic or silicon biochip (commonly known as a genome chip, DNA chip or gene array). Thousands of them can be placed in known locations on a single DNA microarray. (ii)The alternative bead array is a collection of microscopic polystyrene beads, each with a specific probe and a ratio of two or more dyes, which do not interfere with the fluorescent dyes used on the target sequence.
DNA microarrays can be used to detect DNA (as in comparative genomic hybridisation), or detect RNA (most commonly as cDNA after reverse transcription) that may or may not be translated into proteins. The process of measuring gene expression via cDNA is called expression analysis or expression profiling.
Sample Questions
- 1) Define what in DNA polymorphism and its forms?
- 2) Explain how mtDNA polymorphism helped in understanding modern humans in Indian subcontinent?
- 3) Discuss the Y-chromosome tree?
Short Notes
- 1) Polymerase Chain Reaction?
- 2) Sanger method of DNA sequencing?
- 3) RFLP and its uses?
- 4) Microarray and its usage?