- 3.1 Introduction
- 3.2 Inborn Errors of Metabolism: Genesis
- 3.3 Inborn Errors of Metabolism in Man
- 3.3.1 Phenylketonuria
- 3.3.2 Alkaptonuria
- 3.3.3 Galactosemia and Galactosuria
- 3.3.4 Disorders of Lipid Metabolism: Familial Hypercholesterolemia
3.1 INTRODUCTION
Metabolism broadly refers to the sum total of all the chemical changes occurring in a cell, a tissue or the body, and these chemical changes are performed through organized multistep pathways involving a number of enzymes, proteins and biochemical ingredients. There are several pathways or cycles that intersect with each other forming a cohesive co-ordinated network of chemical reactions. The sum total of chemical reactions performed through different pathways can be classified as either Catabolic (degradative) or Anabolic (Synthetic). The breaking down of complex molecules such as proteins, polysaccharides and lipids etc. to simple molecules such as Co2, NH3 (ammonia) and water is the example of catabolic reaction. The synthetic or anabolic path way refers to the chain of events that lead to the formation of complex products from simple precursors; for example the synthesis of polysaccharide, glycogen from glucose. Investigation of metabolism involves elucidation of complex path ways. Each path way is characterized by action of multienzyme sequences, and each enzyme, in turn, may display important catalytic or regulatory features. As far as Catabolism is concerned, the degradation of complex molecules occurs in three stages and Adenosine Triphosphate (ATP) is released as chemical energy as a degradation of energy-rich molecules. Three stages of Catabolism are: a) Hydrolysis of Complex molecules; b) Conversion of building blocks to simple intermediates c) Oxidation of acetyl CoA. Anabolism refers to the process of formation of complex molecules such as protein, polysaccharides, lipids, nucleic acids from small molecules such as amino acids, sugars, fatty acids, nitrogenous bases etc.
3.2 INBORN ERRORS OF METABOLISM: GENESIS
Sir Archibald Garrod’s Paper on “The incidence of Alkaptonuria : A study of Chemical individuality” published in 1902, was a monumental paper that ushered in a new paradigm so far as the history of human genetics is concerned. The publication of the paper by Garrod opened up new perspectives for two reasons. First, the results obtained by Garrod on genetics of Alkaptonuria reinforced the application of Mendel’s paradigm on humans. Second, the findings of the paper subsequently led to the emergence of a new concept as well as an important area of study in human genetics under the rubric “Inborn Error Metabolism”. With the accumulation of evidences on genetic basis of enzyme action in man, the study of inborn errors of metabolism in humans has become an important area of study in human bio-chemical genetics having tremendous application value in medical / clinical genetics.
Garrod’s work gave a strong ground in favour of the concept of one gene-one enzyme hypothesis, which was established in the 1940s and 1950s by the pioneering work of Beadle and Tatum, who were awarded Nobel Prize.
Principles of Inborn – errors of Metabolism
Metabolism broadly refers to complex bio-chemical pathways functioning as an integrated system of chemical reactions controlled in some manner by genes and these genes regulate or control specific reactions in the system by acting directly as enzymes or by determining the specificities of enzymes, where as inborn error metabolism denotes inability on the part of humans to catalyze certain metabolic pathways completely due to alterations of enzymes, and this modification of the action of enzymes is attributed to genetic factor or an inborn factor. These defects or errors or blockages in the action of enzymes are due to mutations in the gene/ genes that code for the enzyme/enzymes. Normally, they usually affect only one in a series of iso-enzymes. If more than one iso-enzyme is affected, these variant iso-enzymes may share common polypeptide chain or secondary effects may occur on the structure of enzyme. The deficiency of an enzyme caused by mutation of a gene that is active only in one tissue affects the phenotypes of its carrier in different ways as compared to an enzyme deficiency affecting many tissues. This is analogous to pleiotropic effect of the gene. However, this phenomenon does not always occur in all humans. It may so happen that an enzyme defect that is found in all tissues some time lead to phenotypic anomaly in one tissue only this is due to the fact that defect is ameliorated more early in other tissues.
3.3 INBORN ERRORS OF METABOLISM IN MAN
The following are the four examples for the Inborn Errors of Metabolism in Man. They are Phenylketonuria (PKU), Alkaptonuria, Galactosemia & Galactosuria and hypercholesterolemia.
3.3.1 Phenylketonuria (PKU)
This metabolic disorder was first described by Folling in 1934 in mentally retarded patients, who emitted a peculiar “mousy” odor. PKU is known as one of the best known inborn errors of metabolism of amino acid in humans. The enzymology of PKU established that L-phenylalanine is an essential amino acid with respect to protein synthesis. It is known that only a small proportion of L-phenylalanine is utilized in normal humans for protein synthesis. The larger proportion is oxidized primarily to tyrosine due to action of enzyme phenylalanine hydroxylase. The hydroxylase structurally contains two protein components: 1) one which is labile and found only in the liver. 2) the other with stable feature is found in other tissues. PKU is primarily caused by complete deficiency of action the enzyme – phenylalanine hydroxylase mostly found in liver. The non-action ofthe enzyme phenylalanine hydroxylase leading to phenylketonuria is in fact a recessive mutation in the gene that codes for the enzyme phenylalanine hydroxylase and the gene for PKU is located on chromosome 12. PKU is an autosomal recessive disorder whose frequency in Northern European populations is about 1/10,000 live births. The Pathways of phenylalanine metabolism is presented in figures 3.2 and 3.3 and the pathways of phenylalanine metabolism in normal individuals and in patients with PKU is presented in figure 3.4.
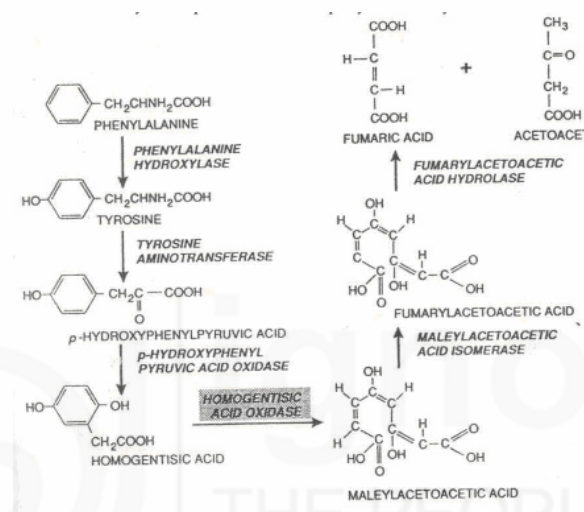
Fig. 3.2: Enzymatic steps in the catabolism of phenylalanine and tyrosine to aceto-acetic acid (Courtesy: Gelehrter, T.D and Collins, F.S. 1998. Principles of Medical Genetics).
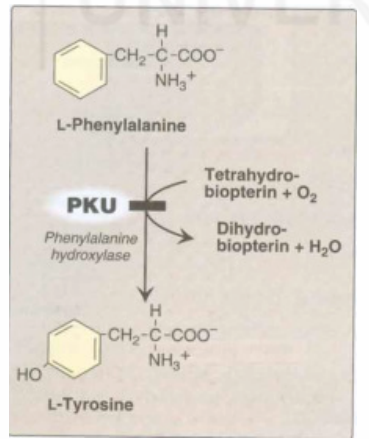
Fig. 3.3 : A deficiency in Phenyllalanine Hydroxylase results in the disease Phenylketonuria (PKU) (Courtesy: Champe, P.C, Harvey, R.A and Ferrier, D.R (Ed). 2005. Biochemistry).
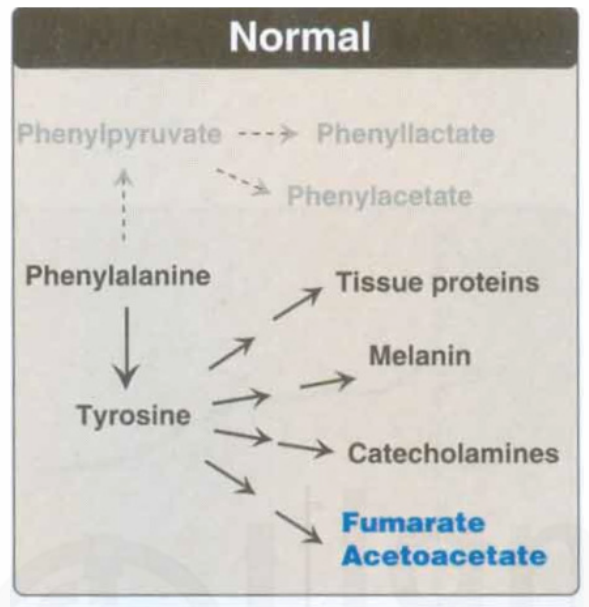
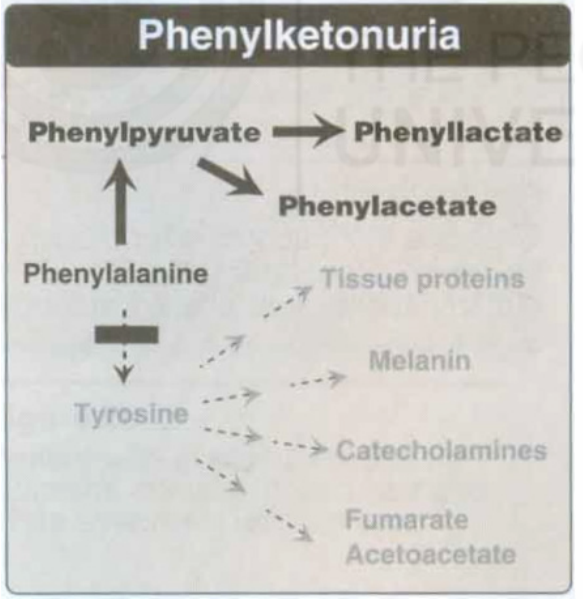
Fig. 3.4: Pathways of phenylalanine metabolism in normal individuals (above) and in PKU patients (below) (Courtesy: Champe, P.C, Harvey, R.A and Ferrier, D.R(Ed). 2005. Biochemistry)
Characteristics of PKU
a) Elevated phenylalanine
The phenotypic–clinical feature of patients with PKU is characterized by microcephaly (small head) and profound mental retardation. As stated above the genetic mutation that blocks the action of the enzyme phenylalanine hydroxylase in the hepatic system leads to low concentration of tyrosine and high concentration of phenylalanine in plasma. The concentration of phenylalanine is elevated in tissues, plasma and urine in persons with PKU. The reason is that three types of metabolites of phenylalanine such as phenyl lactate, phynyl acetate, and phenyl pyruvate, which are not formed in normal individuals due to action of enzyme phenylalanine hydroxylase, are produced in elevated concentration in PKU patients. Thus the name of the disease phenylketonuria (PKU) which signifies the presence of phenyl ketone in the urine in the form of phenyl pyruvate. Such high concentration phenylalanine metabolites such as phenyl-ketones are excreted through urine, giving a characteristic ‘Mousy’ odor. The high concentration phenylalanine in plasma is attributed to mental retardation among infants. It is known that the formation of tyrosine from phenylalanine is catalyzed by the action of enzyme phenylalanine hydroxylase. The reaction requires molecular oxygen and coenzyme – tetrahydrobiopterin (BH4) which is synthesized by the body. One atom of molecular oxygen becomes the hydroxyl group of tyrosine, the other atom is reduced to water. During reaction teatrahydrobiopterin is oxidized to dihydro hydrobiopterin in a separate reaction needing NADPH. Hyper-phenylalanimenia may also caused by deficiencies in the formation of enzyme, that synthesizes or reduces the hydroxylases co-enzyme tetrahydrobiopterin.
b) CNS Syndrome: Mental retardation, failure to walk, talk, seizures, hyperactivity, tremor, microcephaly and failure to grow are characteristic findings in PKU.
c) Hypo-pigmentation: Patients with PKU with high levels of phenylalanine demonstrate a deficiency of pigmentation (fair hair, light skin colour, and blue eyes).
Treatment of PKU : As stated earlier, the PKU is a disease of metabolic disorder, caused by an autosomal recessive mutation, that inhibits the action of enzyme which is responsible for catabolizing phenylalanine to tyrosine. Since PKU has a genetic basis, both prevention as well as treatment of patients of varying age groups both at family level and community level are regarded as public health problem. The most pressing need is to address the issue of rise in phenylalanine metabolites in urine along with high incidence in the serum phenylalanine level. The therapy that was first administered for reducing phenylalanine intake through dietary supplementation was made by Bickel in 1953. Since then, several groups attempted on dietary supplementation programme and have achieved success. The evidence is now confirmed that dietary therapy used have the most ameliorating effect on the development of PKU patients. Two important guidelines have to be borne in mind while treating PKU.
i) In order to prevent brain damage the diet should be started as soon as possible within the first week of life.
ii) The metabolic status of the child especially phenylalanine levels should be monitored carefully.
Adults do not require lifelong treatment since adult frames are resistant to rise to metabolite concentration found in PKU.
3.3.2 Alkaptonuria (AKP)
Alkaptonuria is a rare metabolic disorder involving a deficiency in the enzyme “homogentisic acid oxidase”, which is in fact responsible for the formation of maleylacetoacetate from Homogentisate. The bio-chemical pathway involving the Alkaptonuria is shown in figure 3.1.
Sir Archibald Garrod’s first monumental study on Alkaptonuria in 1902 brought about a new perspective in human genetics. He could establish through his seminal paper that metabolic disorders have genetic basis. This was the first example that genes may register their effect by coding for enzymes genetically. Alkaptonuria was thus recognized as the first autosomal recessive human disease that emanates from the deficiency of an enzyme. The characteristic symptoms of the disease are:
a) Formation of homogentisic aciduria: In this condition, the patients urine contains elevated levels of homogentisic acid, which is oxidized to a dark pigment on standing leading to black colour (fig. 3.5)
b) Development of Arthritis, particularly in the spine and large joints due to the degenerative changes.
c) The deposition of endogenous homogentisic acid autooxidation products in the cartilage and collagenous tissue lead to blue or black ochronotic pigmentation of cartilage, and black deposits in the sclera of the eyes, other features of this condition can include heart problems, kidney stones and prostrate stone.
How common is AKP?: This condition is rare, affecting I in 250,000 to 1 million people world wide. AKP is more common in certain Areas of Slovakia (1 in 19,000) and in the Dominican Republic.
Genetics of Alkaptonuria : It is now clearly established that mutation in HGD gene causes Alkaptonuria. The gene encoding an enzyme called homogentisate 1, 2- dioxgenase (also known as homogentisate oxidase). The gene for HGD is located on chromosome 3 in region 3q21-q23.
The enzyme Homogentisate Oxidase is primarily active in the liver and kidney. It is chiefly responsible for breaking down the amino acid phenylalanine and tyrosine, into a molecule called homogentisic acid. Homogentisic acid oxidase adds two oxygen atoms to homogentisic acid converting homogentisic acid into another molecule called Maleylacetic acetate.
More than 40 mutations in HGD gene have been detected in people with Alkaptonuria. The substitution of the amino acid valine for methionine at position 368 is the most common form of HDG mutation in European population. The mutation in this gene inactivates the enzyme. Without HGD normal gene functioning, the enzyme homogentisic oxidase become ineffective accounting for building up homogentisic acid in the body. The gene for Alkaptonuria is inherited as autosomal recessive trait. The person with Alkaptonuria inherits both copies of the recessive genes from parents who are heterozygotes for Alkaptonuria.
3.3.3 Galactosemia and Galactosuria
Galactosemia & Galactosuria are identified as disorders of carbohydrate metabolism especially of Galactose metabolism.
The major source of Galactose is obtained from lactose – the principal source of milk and milk products. However, some amount of galactose can be obtained by lysosomal degradation of complex carbohydrates such as glycoproteins, and glycolipids – the membrane components of tissues.
Metabolic Pathways: The metabolic pathways that involve metabolism of Galactose are as follows:
- a) Phosphorylation of Galactose
- b) Formation of UDP-galactose
- c) Use of UDP-galactose as a carbon source for glycolysis or gluconeogenesis
- d) Role of UDP -Galactose in biosynthesis reaction.
Galactosemia and galactosuria arise in individuals who are deficient in secreting enzymes– a) Galacto kinase that metabolizes Galactose to Galactose 1-P phosphate. b) Galactose 1. Phosphate Uridyl transferase that converts Galactose1-Phosphate to UDP-Galactose and Glucose1-Phosphate. Galactosuria: As stated earlier the cases of galactosuria and galactosemia are due to the deficiency of galactokinase enzyme that converts galactose to galactose1phosphate.
Classic Galactosemia: It is in fact a deficiency of the enzyme Uridyltransferase. The deficiency is due to autosomal recessive disorder. It causes both galactosemia and galactosuria . The frequency of this deficiency is 1 in 23,000 births. Individuals with Uridyl transference enzyme deficiency facilitate the accumulation of Galactose 1-Phosphate, and consequently excess of galactose in the body lead to vomiting, diarrohea and jaundice. In some cases, the excess accumulation of galactose 1-Phosphate also causes liver damage, severe mental retardation and cataract. Genetically this disorder is an autosomal recessive mutation in the gene that codes for the enzyme Galactose 1-Phosphate uridyl transferase.
3.3.4 Disorders of Lipid Metabolism: Familial Hypercholesterolemia
Familial Hypertension (FH) is an important cause of heart disease. It is one of the most common autosomal dominant disorder. In the individuals with FH the plasma cholesterol levels are twice higher than normal (i.e. about 300-400 mg/ dl). The high cholesterol level is due to deficient or defective LDL receptors leading to enhanced levels of endogenous cholesterol synthesis.
All cells require cholesterol as a component of their plasma membrane. They can either synthesize their own cholesterol or they get it from the extra cellular environment, where it is carried principally by low density lipoprotein (LDL). In a process known as endocytosis (fig. 3.7) LDL-bound cholesterol is taken into In case of homozygotes (cholesterol levels raising from 600 to 1200 mg/dl) the distinctive cholesterol deposits in the skin and tendons called xanthomas occur.
Sample Questions
- 1) Discuss on example of error of amino acid metabolism. Can you treat it by any means?
- 2) Give a brief note on an example of error of lipid metabolism leading to heart disease.
- 3) What is the difference between galactosaemia and galactosuria? Comment
- 4) Write a note on the first metabolic disorders described by Garrod?