- 1.1 Biochemical Genetics
- 1.1.1 History
- 1.1.2 Garrod’s Inborn Errors of Metabolism (IEM)
- 1.1.3 Beadle and Tautum – Genetic Analysis Led to the One-Gene-One Enzyme Hypothesis
- 1.1.4 Genetic Code
- 1.1.5 Revisiting the Question about what is a Gene?
- 1.2 Biochemical or Metabolic Diseases
- 1.2.1 Disorders of Amino Acid Metabolism
- 1.2.2 Disorders of Branched Chain Amino Acid Metabolism
- 1.2.3 Urea Cycle Disorders
- 1.2.4 Disorders of Carbohydrate Metabolism
- 1.2.5 Disorders of Steroid Metabolism
- 1.2.6 Disorders of Lipid Metabolism
- 1.2.7 Lysosomal Storage Disorders
- 1.2.8 Lipid Storage Diseases
- 1.2.9 Disorders of Purine/Pyramidine Metabolism
- 1.2.10 Disorders of Copper Metabolism
- 1.3 Human Blood Group Substances
- 1.3.1 Introduction to Human Blood Groups
- 1.3.2 The ABO Blood Group System-History
- 1.3.3 ABO Blood Groups Substances
- 1.3.4 Antigens and Antibodies
1.1 BIOCHEMICAL GENETICS
In the light of the understanding of basic aspects of genetic variation, let us now move on to one of the very important specialty in human genetics – known as Biochemical Genetics.
- History
Gregor Mendel (1822 – 1884) (Fig. 1.1) is well known as the founder of modern genetics. He conducted breeding experiments from 1856 to 1863 and presented his results in 1865. Using the different varieties of pea plants interms of the flower colour, the seed coat type and many such simple characteristics carefully chosen as the varieties, he analysed the pattern of transmission of these traits in subsequent generations. This led to the discovery of principles of heredity which is now known as Mendelian Inheritance in his honour.
The impact of this research, appeared to be initially minimal and no one realized that Mendel has discovered the basic principles of inheritance. Unfortunately, the work remained dormant for nearly 44 years.
In 1900, three botanists Hugo de Vries, Erich von Tschermark, and Karl Correns did similar experiments with plants and arrived at conclusions similar to those of Mendel.
1.1.2 Garrod’s Inborn Errors of Metabolism (IEM)
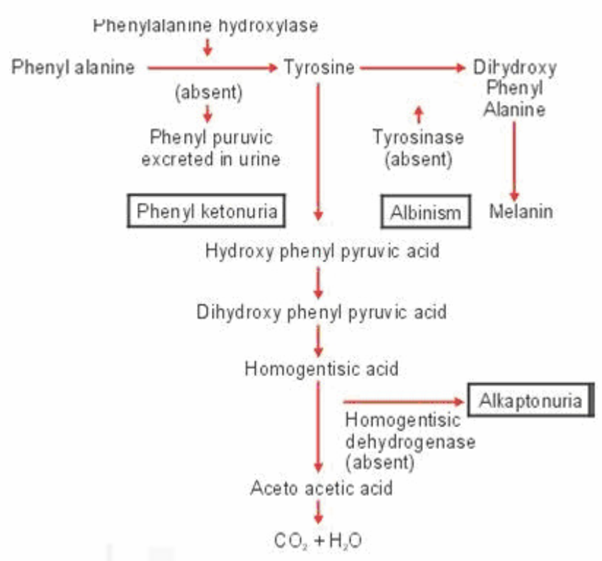
At the time that Mendel’s work was being discovered, English Physician and biochemist Archibald Garrod was studying a rare condition (incidence of 1 in 250,000) known as alkaptonuria (uria means “in the urine”) otherwise healthy new born with the disorder, usually had one striking feature. Their urine upon standing in contact with air, turned black. In their later years, affected individuals show darkening of portions of ears, whites of eye, cartilage and tendons. One to the deposits in the cartilage they suffer with arthritis. Males develop black stones in the prostrate at later age). The dark substance in the urine was the oxidation product of alkapton now called homogentisic acid, which contains a six- carbon ring structure. Other researchers found that the excretion of homogentisic acid in urine was increased, when people with alkaptonuria were fed excess protein or the amino acid tyrosine and phenylalanine, both of which contain benzene rings. Yet individuals who have no alkaptonuria never excrete any homogentisic acid in urine.
Garrod suggested that homogentisic acid is an ordinary product of metabolism that is broken down in normal people but is not degraded in people with Alkaptonuria. This metabolic block results in homogentisic acid being excreted intact in the urine. More specifically, Garrod thought that people with alkaptonuria do not properly metabolize ring- containing fractions of proteins because they lack a special enzyme that is present in unaffected individuals. He proposed that during the normal break down of proteins, phenylalanine is converted to tyrosine, which in turn is changed to homogentisic acid and then simpler to complex products.
Garrod’s analysis did not stop with the concept of metabolic blocks, however. The brilliance of his proposal lay in the genetic connection that he and population geneticist William Bateson were able to make. Garrod noted that alkaptonuria tended to occur in several sibs of a family whose parents were unaffected and also that many affected children were the offspring of first-cousin marriages. This pattern of inheritance fit the requirements for a rare recessive trait, the first human application of Mendel’s newly discovered laws.
Garrod classified Alkaptonuria and three other conditions that he called as Inborn Errors of Metabolism. The four canonical inborn errors described by Garrod are Albinism, Alkaptonuria, Cystinuria and Pentosuria. He suggested that these four genetic conditions must represent only a tiny fraction of the errors of metabolism that exist in human populations, pointing out that many of them may produce no obvious externally manifesting phenotypic effects.
Archibald Garrod (Fig. 1.2) served from 1915 to 1919 as a colonel in the British Army Medical Services in Malta. Then although Garrod’s medical career was extremely successful and distinguished, his pioneering insights into the nature of metabolic disorders and the principle of biochemical individuality was not appreciated during his life time.
Garrod an extraordinary physician scientist made discoveries that were ahead of his time and died long before, we would appreciate fully the information, the knowledge and the wisdom he had passed on to us. Garrod belonged to the period 1857 to 1936. During his lifetime the natural sciences changed our view of the world, and genetics began its journey to the double helix.
Thus was born the Garrod’s bold hypothesis of “inborn errors of metabolism” with its far- reaching assumption that genes were there to produce chemical catalysts, one gene to each highly specialized catalyst.
Garrod the initial founder of human “Biochemical Genetics” belonged to a unique community ahead of his times as rightly appreciated by Scriver (2001) in his felicitation lecture, that Garrod belonged to the clinical-scientic community. He introduced a paradigm new for its day in medicine: he thought that biochemistry is dynamic and different from the static nature of organic chemistry. So this led him to think about metabolic path ways and heredity could explain an “inborn error of metabolism”. During Garrod’s time, he had no idea about the nature of gene. Genes are now well understood: Genomes are being described for Homo sapiens and it is understood that genomes “speak biochemistry ( not phenotype)” Scriver (2011).
1.1.3 Beadle and Tautum – Genetic Analysis Led to the One Gene-One Enzyme Hypothesis
Garrod’s ideas, like Mendel’s were largely ignored for over thirty years although Garrod published a book and several papers on inborn errors of metabolism. The concepts have to be rediscovered by the American geneticists in their influential experiments connecting genes with enzymes. This was carried out in 1940s by George W. Beadle and Edward L. Tautum using a filamentous fungus called Neurospora crassa commonly called red bread mould. They chose this organism because both genetic and biochemical analysis could be done with ease.
In these experiments they identified new mutations that each caused a block in the metabolic pathway for the synthesis of some needed nutrients and showed that each of these blocks corresponded to a defective enzyme needed for one step in the pathway. The experimental approach now called genetic analysis was important because it solidified the link between the genetics and biochemistry.
N. crassa grows in the form of filaments on a great variety of substrates including laboratory medium containing only inorganic salts, a sugar and one vitamin. Such a medium is called as minimal medium because it contains only the nutrients that are essential for growth of the organism. The filaments consists of a mass of branched threads separated into interconnected, multinucleate compartments allowing free interchange of nuclei and cytoplasm. Each nucleus contains a single set of seven chromosomes. Beadle and Tatum recognized that the ability of Neurospora to grow in minimal medium implied that the organism must be able to synthesize all of the other small molecules needed for growth such as amino acids. If the biosynthetic pathways needed for growth are controlled by genes, then a mutation in a gene responsible for synthesizing an essential nutrient would be expected to render a strain unable to grow unless the strain were provided with the nutrient.
The classic experiments of Beadle and Tautum thus showed that the relationship is usually remarkably simple: one gene code for one enzyme. The pioneering experiments united genetics and biochemistry, and for the “one gene one enzyme” concept Beadle and Tautum were awarded a Nobel prize in 1958.
Beadle and Tautum were fortunate to study metabolic pathways in relatively simple organism in which each gene specifies a single enzyme, a relation often called the one enzyme hypothesis.
1.1.4 Genetic Code
In 1953 James Watson and Francis Crick solved the structure of DNA and identified the base sequence as the carrier of genetic information. But the way in which the base sequence of DNA specifies the amino acid sequence of proteins (the genetic code) was not obvious until 10 years. Three nucleotides are necessary to specify a single amino acid. This is the basic unit of the genetic code. So the set of basis that encode a single amino acid is called as a codon. Using limitations in bacteriophage Francis Crick and his colleagues confirmed in 1961 that the genetic code is a triplet code in which three nucleotides encode each amino acid in a protein. Proteins are very important to all living processes; they are in some cases enzymes, which are the biological catalyst and conduct the chemical reactions of the cell. In other cases proteins are structural components of a cell like muscle, nail, lens in the eye. Some proteins help in the function of transporting substances others help in the regulation of vital pathways, communication between cell to cell or in defending a cell from external threat.
Every protein is composed of amino acids, aligned end to end. Twenty common amino acids are known to constitute different proteins. Each amino acid has a central carbon atom bonded to an amino group (NH3+), a hydrogen atom ,a carboxyl group (COO-)and an R (Radical) group. The Radical group differs for each amino acid. The amino acids in proteins are joined together by peptide bonds.
Thus it is very important for you to remember some important concepts like
- 1) The products of many genes are proteins whose action produce the traits encoded by these genes. Proteins are polymers consisting of amino acids linked by peptide bonds. The amino acid sequence of a protein is its primary structure.
- 2) This structure folds to create the secondary and tertiary structures; two or more polypeptide chains may associate to create a quaternary structure.
- 3) The genetic code is a triplet code in which three nucleotides encode each amino acid in a protein.

Because we now know that some enzymes contain polypeptide chains encoded by two (or occasionally more) different genes, a more accurate statement of the principle is “one gene, one polypeptide”.
1.1.5 Revisiting the Question about what is a Gene?
After studying the developments of Genetics as it unfolded above you may be able to appreciate in a nut shell that:
- i) The Mendelian concept of a gene views it as a discrete unit of inheritance that affects phenotype.
- ii) Morgan and his colleagues assigned genes to specific loci on chromosomes.
- iii) We can also view gene as a specific type nucleotide sequence along a region of a DNA molecule as put forth by Watson and Crick.
- iv) We can define a gene functionally as a DNA sequence that codes for a specific polypeptide chain.
Even the one gene one polypeptide definition must be refined and applied selectively, because:
- i) Most eukaryotic genes contain large introns that have no corresponding segments in polypeptides.
- ii) Promoters and other regulatory regions of DNA are not transcribed either, but they must be present for transcription to occur.
- iii) Our definition must also include the various types of RNA that are not translated into polypeptides.
Hence a gene is a region of DNA whose final product is either a polypeptide or an RNA molecule.
1.2 BIOCHEMICAL OR METABOLIC DISEASES
Now we will move on to consider single gene biochemical diseases. The spectrum of disorders known under this category is vast (200 inborn errors of metabolism are known) and a flavor of this fascinating branch of genetic medicine will be given. The metabolic disorders can be grouped based on either the metabolite, metabolic pathway, function of the enzymes or cellular organelle involved.
Only a few IEM are inherited in an autosomal dominant manner. This is because the defective protein in the most inborn errors is an enzyme which is diffusible, and there is usually sufficient residual activity in the heterozygous state for the enzyme to function normally in most situations. If, however, the reaction catalyzed by an enzyme, is rate limiting or the gene product is part of a multimeric complex, the disorder can manifest in the heterozygous state, i.e., be dominantly inherited.
1.2.1 Disorders of Amino Acid Metabolism
The best known under this category are phenylketonuria (PKU), alkaptonuria, occulocutaneous albinism, homocystinuria, maple syrup urine disease.
The untreated 11 year old boy (left) is severely retarded. His two and half year old sister (right) who was treated from early infacy with a low phenylalanine diet, has normal intelligence. Phenylketonuria can lead to severe brain damage and mental retardation.
1.2.2 Disorders of Branched Chain Amino Acid Metabolism
The essential branched chain amino acids leucine, isoleucine and valine have a part of their metabolic pathways in common like the previous set mentioned above. Maple syrup urine disease comes under this category. It is an autosomal recessive disorder that presents with vomiting in the first week of life followed by death within a few weeks. There is a specific odour of the urine similar to the maple syrup. This results in the excretion of increased branched chain amino acids leucine, isoleucine and valine in the urine. Treatment involves a diet that limits the three branched chain amino acids to the extent that it is necessary only to help the new born to grow. Affected individuals are susceptible to deterioration due to catabolic degradation of proteins.
1.2.3 Urea Cycle Disorders
Urea cycle helps to remove waste nitrogens from the amino groups of amino acids arising from the day to day production of proteins. The sight of action is mainly liver cells and the cycle has a five step metabolic pathway. Deficiencies of enzyme in the urea cycle will result in intolerance of protein due to the accumulation of ammonia in the body. This is also known as hyperammonemia. In normal individuals without enzyme deficiency it converts two molecules of ammonia and one bicarbonate into urea. Excess of ammonia levels are toxic to the central nervous system and it can also result in coma and death when untreated. These disorders are fortunately rare.
1.2.4 Disorders of Carbohydrate Metabolism
Examples are Galactosemia, hereditary fructose intolerance, glycogen storage diseases of liver and muscle.
1.2.5 Disorders of Steroid Metabolism
This is an inborn error in the biosynthetic pathways of cortisol. Due to this disorder new born female infants present with virilization of the external gentalia that is the commonest cause of ambiguous gentalia in female new borns. Affected infants in addition to requiring urgent correct assignment of gender are treated with replacement cortisol along with fludrocortisone if they have salt losing form of defect.
1.2.6 Disorders of Lipid Metabolism
Familial hypercholesterolemia where the person will have raised cholesterol level with high risk of developing coronary artery disease. The high cholesterol level is due to deficient or defective function of LDL receptors leading to increased levels of endogenous cholesterol synthesis.
1.2.7 Lysosomal Storage Disorders
While the previous six types of inborn errors of metabolism were due to enzyme defects leading to accumulation of intermediate metabolic precursors, there are other metabolic disorders due to accumulation of very large polysaccharides. Certain macro molecules which are complex need to be degraded consistently in the cell.
Children born with lysosomal disease are apparently normal at birth but tend to accumulate a variety of macro molecules due to a deficiency of lysosomal enzymes. Examples are Hurler Syndrome, Hunter Syndrome, Sanfilippo Syndrome, Morquio syndrome, all put together known as Mucopolysaccharidoses (MPS).
Face of a male with mucopolysaccharidosis. Note the characteristic coarsening of their facial features.
1.2.8 Lipid Storage Diseases
Tay-Sachs disease is the best known lipid storage diseases where in there is a progressive deposition of lipid or glycolipid primarily in the liver and spleen. Central nervous system involvement results in progressive mental deterioration, deafness, visual impairment, spasticity and progressive rigidity . 1 in 3600 persons of Ashkenazi Jewish ancestry are known to be affected with this disorder.
1.2.9 Disorders of Purine/Pyramsidine Metabolism
Lesch-Nyham Syndrome is a good example. Accumulation of excessive amounts of uric acid and some of its metabolic precursors. The main effect on the central nervous system resulting in uncontrolled movements, spasticity, mental retardation and compulsive self- mutilation.
1.3 HUMAN BLOOD GROUP SUBSTANCES
1.3.1 Introduction to Human Blood Groups
The red blood cells of human blood contain a great many antigens mostly proteins and according to the presence of antigens, human blood can be classified into different blood group systems, e.g ABO blood group, MN blood group, RH blood group. All of these blood groups in man are under genetic control, each blood group system being under the genetic control of genes at a single locus or of genes that are closely linked and behave in heredity as though they were in single locus.
1.3.2 The ABO Blood Group System- History
Blood has two main components: Serum and cells. Karl Landsteiner noted that the sera of some individuals caused the red cells of others to agglutinate. This observation led to the discovery of the ABO blood group systems. Based on the reactions between the red blood cells and the sera, he was able to divide individuals into three groups: A, B, and O. Two years later, two of his students discovered the fourth and rarest type, namely AB. Landsteiner described A, B, and O groups; Alfred von decastello and Adriano sturli discovered the fourth type, AB, in 1902.
Karl Landsteiner was born June 14, 1868 in Vienna, Austria. In 1897 he pursued his interest in the emerging field of immunology and in 1901 published his discovery of the human ABO blood group system. He won the 1930 Nobel Prize for Physiology or Medicine for his discovery of the major blood groups and the development of the ABO system of blood typing that has enabled blood transfusions.
Due to less developed communication system in those days, it was subsequently found that Czech serologist Jan Jansky had also independently pioneered the classification of human blood into four groups, although his name is not so much talked about, except in Russia and former states of USSR around that time in America, Moss published a similar work on blood group in 1910, from USA.
Ludwik Herzfeld and von Dungern discovered the heritability of ABO blood groups in 1910–11. Felix Bernstein earns the credit for studying the blood group inheritance pattern to be due to, multiple alleles at one locus in 1924.
1.3.3 ABO Blood Groups Substances
Watkins and Morgan, in England, discovered that the ABO epitopes were conferred by sugars, to be specific:
- i) N-acetylgalactosamine for the A-type
- ii) Galactose for the B-type.
Published literature states that the ABH substances are all attached to glycosphingolipids – a long polylactosamine chain that contains the major portion of the ABH substances attached to it.
Later, Yamamoto’s group showed the precise glycosyl transferase designates the A, B and O epitopes. Epitope is the antigenic determinant on an antigen to which the paratope (the specific site in the immunoglobulin) on an antibody binds.
The genetic basis of the ABO blood group system is an example of multiple alleles. There are three alleles, A, B, and O, at the ABO locus on chromosome 9. The expression of the O allele is recessive to that of A and B, which are said to be co-dominant. Thus, the genotypes AO and AA express blood type A, BO and BB express blood type B, AB expresses blood type AB, and OO expresses blood type O. In the past, ABO blood group typing was used extensively both in forensic cases as well as for paternity testing.

The ABO blood group system is an important blood group system to be considered during human blood transfusion. The associated anti-A antibodies and anti-B antibodies are usually IgM antibodies, which are usually produced in the first years of life by sensitization to environmental substances such as food, bacteria, and viruses. ABO blood types are also present in apes such as chimpanzees, bonobos, and gorillas.
Thus ABO group substances are distinct, genetically determined group of human erythrocyte antigens represented by two blood factors (A and B) and four blood types (A, B, AB, and O).
1.3.4 Antigens and Antibodies
Blood grouping, is essentially based on the definition of antigen and antibody. An antigen is a substance, usually a protein or a glycoprotein, when injected into a human (or other organism) that does not have the antigen, will cause antibody to be produced. Antibodies are a specific type of immune-system proteins known as immunoglobulins, whose role is to fight infections by binding themselves to antigens. In the case of the ABO blood groups, the antigens are present on the surface of the red blood cell, while the antibodies are in the serum. These antibodies are unique to the ABO system and are termed “naturally occurring antibodies.” The table shows the relationships between blood types and antibodies.
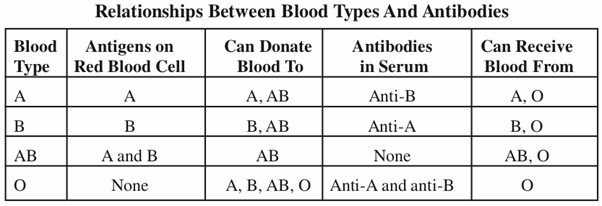
ABO blood group-matching is very important in transfusion. Blood group O individuals are said to be universal donors, because their blood can be used for transfusion in individuals who have any one of the four blood types. On the other hand, individuals with blood type A can only donate to either type A or type AB, and individuals with blood type B can only donate to B or AB types. AB individuals can only donate to type AB. However, before any transfusions, donor blood is mixed with serum from the recipient (a process called cross matching) to ensure that no agglutination will occur after transfusion.
Sample Questions
- 1) Describe with example some classic examples of inborn errors of metabolism.
- 2) Discuss the concept of gene in the light of Beadle and Tautum’s work.
- 3) Define the basis of human ABO blood group systems