Deletion
A deletion refers to the loss of a segment of a chromosome. This leads to the loss of the genes present in the missing region. A single break in the chromosome leads to the loss of the terminal segment and is called terminal deletion . Intercalary deletion, however, involves two breaks in the chromosome, loss of the segment, and rejoining of the two chromosomal parts. Very large deletions are usually lethal because the monosomic condition of the large number of genes of the missing fragment reaches the level of genetic imbalance that cannot sustain life. Usually any deletion resulting in loss of more than 2% of the genome has a lethal outcome. Microdeletions, however, are reported and documented for specific disorders.
Cri-du-chat Syndrome
This syndrome results from a deletion on the short arm of chromosome 5. It is also known by other names such as 5p deletion syndrome and Lejeune’s syndrome. The disorder gets its name from the characteristic cat-like cry of affected infants. Described first by Jérôme Lejeune in 1963, this disorder has an incidence of 1 in 25,000 live births. This disorder, being autosomal, should affect males and females in equal frequencies; but incidence is seen to be more in females by a ratio of 4:3 of females: males affected.
The deletion occurring on the short (p arm) arm of chromosome 5 varies in different affected individuals. The phenotypic effects are also shown to vary between individuals. Most cases show deletion of 30 to 60% of the terminal region of the short arm. Studies show that larger deletions tend to result in more
severe intellectual disability and developmental delay than smaller deletions. The chromosome 5 pair from a karyotype of an individual with this syndrome.
Affected individuals characteristically show a distinctive, high-pitched, catlike cry in infancy with growth failure, microcephaly, facial abnormalities, and mental retardation throughout life. Some common clinical manifestations are:
• Cry that is high-pitched and sounds like a cat
• Downward slant to the eyes
• Low birth weight and slow growth
• Low-set or abnormally shaped ears
• Mental retardation (intellectual disability)
• Partial webbing or fusing of fingers or toes
• Slow or incomplete development of motor skills
• Small head (microcephaly)
• Small jaw (micrognathia)
• Wide-set eyes
Duplications
Duplications, like deletions, can cause abnormal phenotypic effects. They usually arise by errors in homologous recombination (unequal crossing-over).
Duplications have their importance not only in medical genetics, but also in evolutionary genetics. The presence of an extra copy of the gene virtually makes it free of selection pressure. Thus, it contributes to diversification of protein functions resulting in families of proteins. Proteins of such families have related
functions differing in the task they are specialized for. A classic example is that of the globin genes. Different globin proteins express during different times of development, each of which is specialized to transport oxygen under those conditions. These differences arose by gene duplications. Without digressing too much we shall now look at the clinical significance of duplications exemplified by Charcot–Marie–Tooth disorder.
Charcot–Marie–Tooth (CMT) Disorder
This disorder results from duplication in the short arm of chromosome 17 in the region 17p12. It is a hereditary motor and sensory neuropathy that affects the nerve cells of the individual. Affected individuals typically show loss of touch sensation and muscle tissue. The chromosomal basis of this disorder is varied, with 17p12 duplication being one of the causes. The severity and symptoms shown differ depending on the region affected, and the presence of other chromosomal abnormalities associated with the duplication. In CMT type 1A, the duplication causes more of the protein to be produced from the genes in that region. This causes the structure and function of the myelin sheath around nerve fibres to be abnormal, causing various clinical manifestations such as:
• Weak feet and lower leg muscles
• Foot deformities (eg: high arch)
• Difficulty with fine motor skills due to muscle atrophy
• Mild to severe pain as age progresses
• May lead to respiratory muscle weakness
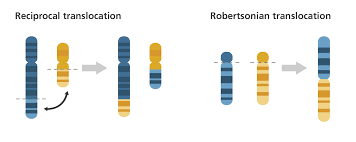
Robertsonian Translocation
Translocations generally do not result in loss of genetic material. Robertsonian translocations, however, result in the loss of small parts of the chromosomes involved. The fusion of two acrocentric chromosomes with the subsequent loss of the two short arms is termed Robertsonian translocation or centric fusion . Although this translocation causes loss of the short arms, it is maintained as a balanced translocation. This is explained by the fact that the genes on the short arms are most rRNA genes that are present in many copies on other chromosomes; thus deletion of these copies doesn’t have much phenotypic manifestation as you might expect.
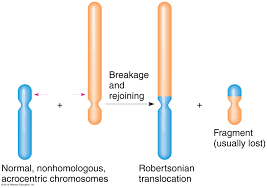
One of the commonly seen such translocation is between chromosome 14 and 21, that gives rise to individuals showing characteristics of Down syndrome. Since this translocation is functionally a balanced translocation, individuals with this aberration usually do not show any abnormal phenotype. Their effects are only seen in the next generation due to production of abnormal gametes. Balanced changes cause disturbances during the meiotic segregation. Due to this, the resulting gametes end up with loss of chromosome, or gain chromosome.
Reciprocal Translocation
Reciprocal translocation involves breaks in two chromosomes and the subsequent exchange of segments between the two chromosomes . Reciprocal translocation do not change the number of chromosomes. However, they may change the size and type of chromosome if the segments being exchanges are differing in size . For reasons not yet clear, reciprocal translocations involving chromosomes 11 and 22 are fairly common in the population.
Reciprocal translocations can give rise to deletion-duplication conditions and can cause disorders associated with such conditions. However, as in the case of Robertsonian translocation, an individual possessing the translocation himself would not show any abnormal phenotype. Due to disturbances during meiotic segregation of these translocated products, individual of the next generation have a possibility of showing abnormal phenotype.
Inversions
An inversion is a condition wherein a segment of a chromosome is inverted. This is caused by two breaks in the chromosome and the subsequent rejoining in a reverse manner. This changes the order of genes on that chromosome and does not cause any changes in the chromosome number . Depending on the involvement of the centromere, inversion are of two types – pericentric and paracentric.
• Pericentric inversions occur when the inverted segment that includes the centromere. The product after inversion can differ significantly in the arm length and thus change the type of chromosome .
• Paracentric inversions occur when the inverted segment does not include the centromere. The product after inversion remains the same type as the original except for a change in the order of genes .
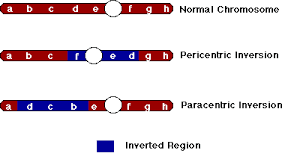
Pericentric and paracentric inversion are both balanced rearrangements because they do not cause any net loss or gain of genes. Except in the very rare cases that the break points in the chromosome is within a gene (which gets disrupted), individuals with inversions do not show any abnormal phenotype. As you may expect, their effects are seen as deletion-duplication only in the next generation.
Isochromosomes and Ring chromosomes
Isochromosomes
An isochromosome is an abnormal chromosome with two identical arms – either having two short (p) arms or two long (q) arms. An isochromosome, thus, has an entire arm deleted along with the duplication of the other arm. This type of aberration is caused due to the transverse separation of the centromere during cell division instead of the normal lateral separation .
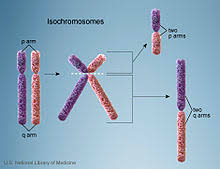
Isochromosomes, due to their deletion-duplication nature, cause abnormal phenotypes in individuals possessing them. The most common isochromosome is that of the X chromosome. This condition leads to the individual showing phenotypic charactertictics of Turner syndrome. The missing genes on the missing arm contributes to the development of Turner syndrome in these females. Such X-isochromosomes account for as much as 15% of Turner syndrome cases.
Ring chromosomes
Ring chromosomes are formed when a chromosome losses its telomere regions and joins back on itself end-to-end. Breaks at the terminal regions cause the chromosome to have “sticky ends” because of loss of telomere region. These end, thus, join with each other causing the chromosome to become circular or ‘ring-like’ . Since the two terminal fragments are lost, loss of genes in those regions can have an effect on the phenotype. If these regions have important genes, their consequences can be serious abnormality in the phenotype.
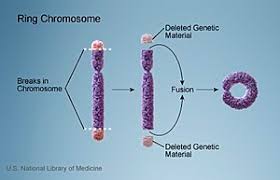
Disorders caused by ring chromosomes are not due to the ring formation itself, but due to the deletion of the genes in terminal regions. Also, ring chromosome are unstable during mitosis, hence the daughter cells may have lost the chromosome altogether. This results essentially in a monosomy. As much as 5% of Turner syndrome cases are shown to be due to ring chromosome-X. Some of the other disorders include ring chromosome 20 syndrome where a ring formed by one copy of chromosome 20 is associated with epilepsy; ring chromosome 14 and ring chromosome 13 syndromes are associated with mental retardation and dysmorphic (malformation) facial features; ring chromosome 15 is associated with mental retardation, dwarfism and microcephaly (small head).
Reasons for Structural Abnormalities
Robertsonian Translocation
As discussed before, Robertsonian translocation or centric fusion, causes a balanced rearrangement in the individual without any phenotypic abnormalities;
however, due to improper meiotic segregation they give rise to trisomy-like and monosomy-like conditions in the offspring of such individuals.
A normal chromosomal complement in humans consists of two copies each of the 22 chromosomes and XY (for males) or XX (for females) – total of 46. Let us consider an individual who has a Robertsonian translocation between chromosomes 14 and 22. This person has a total of only 45 chromosomes. He
has two copies of all the other chromosomes except 14 and 21. For this pair of chromosomes he has one chromosome 14, one chromosome 21 and one translocation 14/21 chromosome.
In a normal individual, during meiosis, one copy of each of these chromosome moves to each pole. This results in daughter cells each containing one copy of 14 and one of 21. In a translocation individual, however, because there are three chromosomes instead of four two of them move to one pole and one moves to another pole. This causes abnormal chromosomal constituents in the daughter cells (gametes). There are different possible ways of these three chromosomes segregating . It is clear that only a small portion of gametes produced by individuals with such balanced translocations can produce normal offspring. Thus, a Robertsonian translocation can give rise to monosomies and trisomies of different chromosomes and their associated syndromes
Although these abnormalities do not cause true trisomies or monosomies, they give rise to conditions that are akin to true trisomies and monosomies. This is because, as stated before, the long arms of these chromosomes contain the bulk of the genes for that chromosome; presence of extra copies of the long arm has the same effect as having an extra copy of the entire chromosome.
Reciprocal Translocation
Translocations not only cause trisomy-like and monosomy-like conditions, they also produce deletion-duplication conditions. A deletion-duplication is a condition where one segment of the chromosome is missing (deletion) and another is present in an extra copy (duplication). Figure 2.19 shows an example of a deletion duplication condition.
Reciprocal translocations, wherein there is a mutual exchange of segments between two chromosomes, cause abnormal meiotic segregation. This abnormality is due to the formation of a quadrivalent of the four chromosomes during pairing (Figure 2.20). This structure is formed because the chromosomal segments always pair with their homologous regions. When such a complex structure is formed, separation of the chromosomes can happen in different ways depending on their orientation in the spindle. Figure 2.20 shows the different possibilities of segregation of a quadrivalent formed from reciprocally translocated chromosomes.
By analyzing the segregation products you should be able to predict the condition of the offspring from such a gamete. The first two segregation patterns produced phenotypically normal offspring. The next two segregation patterns may produce surviving offspring, but they will show abnormal phenotype due to the deletion duplication condition. Depending on the size of the del-dup segment the severity may vary. The last two segregation patterns are usually lethal. This is due to the del-dup segments being very large in these cases. If you recall, deletions of over 2% of the genome is incompatible with survival.
Hence, translocations by themselves do not cause deletions or duplications; it is only in the next generation that their effects are seen. It cannot be emphasized enough that a balanced translocation carrier will most probably have normal phenotype, unless the breakpoint disrupts some gene. As with all balanced rearrangements, we shall see that inversions, too, cause deletions and duplications because of abnormalities in meiotic division.
Inversions
Inversions are balanced genetic rearrangements that invert segments within the chromosome. Depending on the involvement of the centromere they are either paracentric or pericentric . It is important to distinguish between these two types because the crossover products after meiosis is different for
each. Inversions too cause the formation of “inversion loops” during meiotic pairing. Because one of the two homologous chromosomes contains the inversion, it folds back into a loop to allow for maximum homologous pairing . Crossing over is a unique event in meiosis that causes recombination between the homologous pair of chromosomes. When crossing over occurs in a region within an inversion loop, it gives rise to recombinant products that contain deletion and duplication.
The formation of the inversion loop produces maximum homologous regions to be paired up. In pericentric inversions the inversion loop contains the centromere and in paracentric inversion the centromere is outside the inversion loop. Crossing over outside the inversion loop will give rise to normal chromosomes and inversion chromosomes. A crossover within the inversion loop, however, produces two non-recombinants (one normal and one inverted) and two recombinants (that contain deletion and duplication). These recombinants will contain duplication of certain genes along with deletion of other genes.
In pericentric inversions the deleted and duplicated segments do not involve the centromere; hence four types of gametes will be produced. Two of these will contain the aberrations; depending on the extent of the aberration it may or may not be compatible with survival. In paracentric inversions the deleted and
duplicated segments involve the centromere, hence we get one dicentric (containing two centromeres) and one acentric (containing no centromere) chromosome as recombinants. The dicentric chromosome forms a dicentric bridge during anaphase and thus arrests cell division (does not produce a gamete). The acentric chromosome is lost during division and thus doesn’t produce any viable gamete. Hence, only two types of gametes are produced from such individuals – one normal and one containing the inversion. These offspring will have normal phenotype because the inversion itself is a balanced rearrangement. Hence, the inversion itself will tend to persist in the population