- 3.1 Introduction
- 3.2 Human Genome Project (HGP)
- 3.2.1 History of HGP
- 3.2.2 Goals of Human Genome Project
- 3.2.3 Strategies of Sequencing
- 3.2.3.1 Approach of International Consortium
- 3.2.3.2 Approach of Celera Genomics
- 3.2.4 Genome Donors for Sequencing
- 3.2.5 Genome Assembly
- 3.2.6 Genome Annotation
- 3.2.7 Observations Drawn from Human Genome Sequencing
- 3.2.8 How is the Human Genome Arranged or Organised?
- 3.3 Benefits or Applications of Human Genome Project
- 3.3.1 Molecular Medicine
- 3.3.2 Risk Assessment
- 3.3.3 Energy and Environment
- 3.3.4 Anthropology, Evolution and Human Migration
- 3.3.5 Forensic Science
- 3.3.6 Agriculture and Livestock Breeding Drought
- 3.4 Disadvantage of HGP
- 3.5 Post Genomic Era Studies
- 3.6 Need for Individual Diploid Human Genome Sequence
- 3.7 Spin Off of HGP
- 3.7.1 1000 Genomes Project
- 3.7.2 Haplotype map or HapMap
- 3.7.3 Protein Structure Initiative
- 3.7.4 Human Epigenome Consortium
- 3.7.5 Human Genome Diversity Project (HGDP)
- 3.8 Ethical, Legal and Social Implications (ELSI)
What is a genome?
A genome represents total set of different DNA molecules (DNA content) including all of its genes along with spaces between them in an organelle (like mitochondria), cell or an organism. The genome in a species is organised in a specific manner with features to co-ordinate various functions and also reproduction to keep up with continuity of the species. Each genome is a blue print that contains all of the information needed to build and maintain an organism. Human genome is more complex with variation in its organisation found in the nucleus and mitochondrial components. A complete sequence of human mitochondrial (mt) genome was published in 1981 By Fred Sanger and his colleagues.
HUMAN GENOME PROJECT (HGP)
The human genome project was an international effort to sequence every nucleotide in the human genome and to identify all the genes contained within the genome. This effort was coordinated by United States Department of Energy and National Institutes of Health (NIH). It was the highest ever funded programme in biology and laboratories from UK, Japan and Germany were also associated with it.
2 Goals of Human Genome Project
The goals set by Human Genome Project were:
- Identifying all of the estimated 30,000 genes in human DNA and mapping each gene to a site on one of the 23 chromosomes.
- Production of a variety of physical maps of all human chromosomes and that of selected organisms.
- Determination of the complete sequence of human nuclear genome and that of selected model organisms.
- Development of the capabilities for collecting, storing, distributing and analyzing the data generated.
- Creation of necessary technologies to meet the goals of the project.
- Identification of the human genome variation between persons (i.e. single nucleotide variations between any two persons) since such variations are expected to play an important role in individual’s response to infections, drugs and toxins.
- Comparison of human genomes with that of model organisms like bacteria, mouse, yeast, nematode, fruit fly, etc.
- Developing advanced computational capability to collect, store and analyse sequencing data.
- Addressing the ethical, legal and social implications (ELSI) concerned to the use of genetic tools and data.
- Developing interdisciplinary training programmes for future genomics researchers.
3 Strategies of Sequencing
The basis for the human genome sequencing was the dideoxy sequencing method developed by Fred Sanger and his colleagues in 1977. The basic principle of the technique remained the same in HGP programme but with improvements made regarding the efficiency by using the fluorescence labeled automated sequencers and capillary sequencers which helped in obtaining much higher sequencing throughputs (Fig. 3.2a and 3.2b). Dedicated computer programmes like PHRED, PHRAP were developed simultaneously which helped sequence interpretation, scanning for overlapping regions and data assembly.
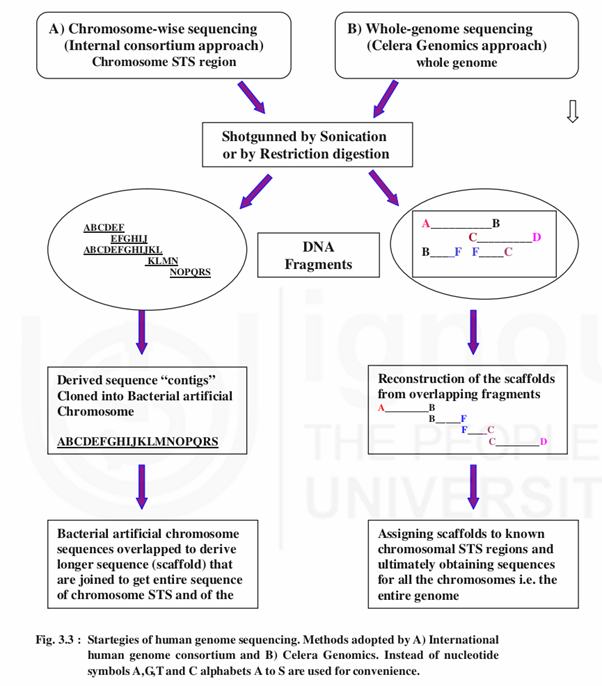
Two different approaches were used to determine the first draft of genome sequence 1) Public funded project planned by International Human GenomeSequencing Consortium (IHGSC) headed by Francis Collins and 2) Planned by the Celera Genomics led by Craig J. Ventor (Fig.3.3).
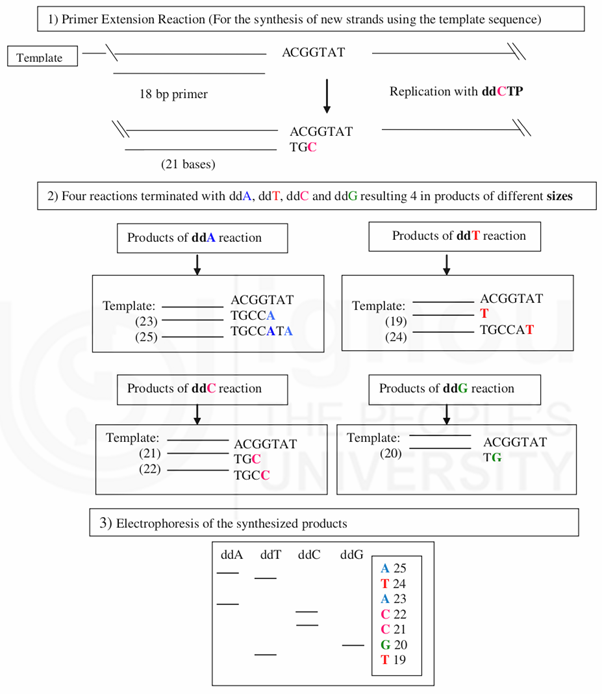
Using the target sequence ACGGTAT, primer with 18 bps, radiolabelled dideoxy nucleotides (ddA, ddT, ddC and ddG) and polymerase enzyme new strands are synthesised. In the presence of dideoxy nucleotides the synthesis of new strands of DNA are terminated whenever the specific ddNTP is added. Thus products of different sizes are generated (that are of 19 to 25 bps length) which can be separated by gel electrophoresis. The original sequence (TGCCGT) can be read from the order/ladder of electrophoresis bands developed on the gel.
3.2.3.1 Approach of International Consortium
The public funded HGP was based on the “hierarchical shotgun” sequencing which involves random cleaving by sonification of starting DNA (from a chromosome) into several hundreds of fragments (150,000 bps in length) followed by end repair. These fragments are then cloned into a vector known as “Bacterial Artificial Chromosomes” or BACs which are derived from genetically engineered bacterial chromosomes. These vectors containing the genes or DNA fragments can be inserted into bacteria where they multiply using the bacterial DNA replication machinery. The BAC contents were known to correspond to specific locations on the chromosomes called sequence tagged sites (STSs). Several copies of each BAC were cut or ‘shotgunned”, into approximately 80 overlapping pieces which were then sequenced. A powerful computer programme was used to assemble the overlapping pieces into overall sequence for each chromosome. This process is nothing but mapping. The entire procedure is referred as “hierarchical shotgun” since the genome is first broken into relatively large pieces which are then mapped to chromosomes before being selected for sequencing.
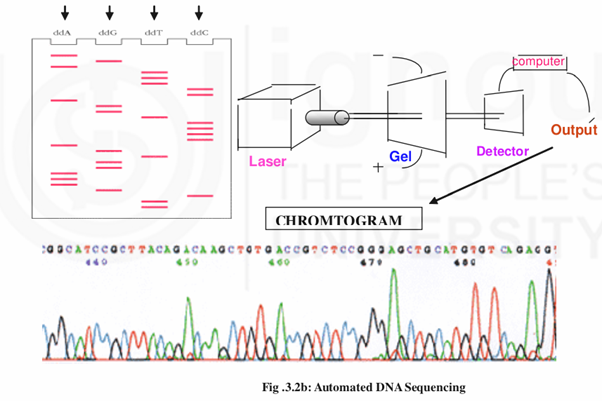
The primer extension reactions carried out in automated sequencing is similar to that of Sanger’s method except that the primers in each reaction are labeled with a different fluorescent staining molecule that emits light of a distinct color i.e. red for thymine, green for adenine, blue for cytosine and black for guanine. The different primer extension reaction products separate according to size upon gel electrophoresis. The bands are color coded. A laser beam that passes through the gel excites the fluorescent tag on each band and the detector analyses the color of the resulting emitted light. This information is converted into a sequence of bases and is stored in a computer. Print outs can be taken from the computer and the chromatogram will give the sequence details as peaks of different colors corresponding to the color of the fluorescence dye used for each base. In theabove diagram the sequence of nucleotides in 440- 446 positions are TCCGCTT that can be read by the color of the peaks.
3.2.3.2 Approach of Celera Genomics
Celera Genomics headed by J. Venter followed “whole-genome shotgun” technique to sequence the human genome employing pairwise end sequencing. This technique was used to sequence bacterial genome of up to 6 million base pair in length, but not for large genome of 3.2 billion base pairs found in human genome. The technique skipped the BAC stage and used shotgunning multiple copies of the genome into small pieces. These pieces were then assembled into large overlapping sequences called “scaffolds” (frame work) using powerful computer programmes. There were 119,000 scaffolds which were assigned to chromosomal sequence tagged sites (STSs). Celera company used information from public database but denied the access to any one to the private database generated by it. Celera’s approach was rapid and of low cost involving only $ 3 millions as compared to publicly funded project of $3.2 billions.
3.2.4 Genome Donors for Sequencing
In the IHGSC, an international public-sector HGP, researchers collected samples of blood from females and that of sperm from males from large number of donors. Only a few of many of these (2 male and 2 female samples out of 20 each) were processed for DNA sequencing. Neither the donors nor the scientists knew the source of the samples and thus identity of the donors were protected. Much of the sequence (>70%) of the reference genome produced by the public HGP came from a single anonymous male donor from Buffalo, New York. For the Celera Genomics private-sector project samples were collected from 21 different individuals and only DNA of 5 individuals were used for sequencing.
3.2.5 Genome Assembly
Genome assembly which is a difficult computation method, is the process of arranging a large number of short sequences of DNA together to create a representation of the original chromosomes from which the DNA originated. In a shotgun sequencing project, all the DNA from an organism is first broken into millions of small pieces. These pieces are then “read” by automated sequencing machines, which can read up to 1000 nucleotides (with the bases adenine, guanine, thymine and cytosine). A genome assembly algorithm picks up all the pieces of DNA and aligns them to one another by detecting all regions where two of the short sequences, or “reads” overlap. These overlapping reads can be merged together, and the process continues. The draft genome sequence is produced by combining the sequenced contigs (ordered arrangement of cloned overlapping fragments) information and using linking information to create “scaffolds” (frame work). Scaffolds are then positioned or assigned to known chromosomal sequences tagged sites (STSs) creating a path.
3.2.6 Genome Annotation
Once the draft sequence is ready, Genome annotation has to be followed. Genome annotation is the process of attaching biological information to the sequences obtained. It is a major challenge for the HGP and covers a) structural annotation that deals with identification of genomic elements like open reading frames (ORFs), gene structure, coding regions and location of regulatory motifs and b) functional annotation that deals with attaching information about biological function, biochemical function, gene regulation and interactions and gene expression to the genomic elements. These steps involve both biological experiments and in silico analysis (bioinformatics).
Automatic annotation tools perform all the annotation by computer analysis.The basic level of annotation is using basic local alignment search tool (BLAST) for finding similarities between the sequences studied and then annotating genomes based on that. Genome annotation is an active area of investigation undertaken by different organisations which publish the results of their efforts in publicly available biological databases accessible via the web and other electronic meansThe HGP catalogued the information on the sequence of nucleotides in thousands of DNA fragments in a public database called GenBank maintained by US National Center For Biotechnology Information (NCBI) and sister organisations in Europe and Japan. From GenBank data base, sequences of known and hypothetical genes and proteins can be retrieved. The data bases are open for every one through internet. Other organisations like, Genome Bioinformatics group from University of California, Santacruz and Ensemble provide additional data and annotation and powerful tools for visualising and searching it.
3.2.7 Observations Drawn from Human Genome Sequencing
The draft genome sequence published in 2001 and the complete genome sequence published in 2004 reported the following findings:
- The human genome contains 3.1647 billion chemical nucleotide bases (A, C, T, and G).
- The total number of genes estimated at 30,000.
- The average gene consists of 3000 bases, but sizes vary greatly, with the largest known human gene being dystrophin at 2.4 million base.
- Almost all (99.9%) nucleotide bases are exactly the same in all people and the functions are unknown for over 50% of discovered genes.
- The number of genes in human beings is of same range found as in mice and round worms. Understanding how these genes express themselves and function would help to know how human diseases are caused.
- About 1.1% to 1.4% of the genome’s sequence codes for proteins that carry out required functions in an organism.
- 98% of the genome is non-coding for proteins and misnamed as “junk DNA”. Now much of the junk DNA is found to code for RNA which regulates other genetic and cellular functions.
- The human genome has high level of segmental duplications i.e. nearly identical, repeated sections of DNA than the other mammalian genomes. These repeated sequences may underline the creation of primate specific genes.
- Repetitive sequences are thought to have no direct functions, but they shed light on chromosome structure and dynamics.
Over time, the repeats reshape the genome by rearranging it, creating entirely new genes, and modifying and reshuffling existing genes. During the past 50 million years, a dramatic decrease seems to have occurred in the rate of accumulation of repeats in the human genome.
3.2.8 How is the Human Genome Arranged or Organised?
Genome sequencing facilitated better understanding of a) Nature of the genes controlling several traits b) Nature of Mutations resulting in altered functions of proteins c) Manipulation of the genome and predicting the consequences.
The human genome has gene-dense “urban centers” that are predominantly composed of the DNA building blocks G and C. In contrast, the gene-poor “deserts” are composed richly of DNA building blocks A and T. Under the microscope GC- and AT-rich regions can be observed as light and dark bands on chromosomes representing euchromatin and heterochromatin regions. Genes appear to be concentrated in random areas along the genome, with vast expanses of long stretches of non-coding DNA between them. Stretches of up to 30,000 C and G bases repeating over and over often occur adjacent to gene-rich areas, forming a barrier between the genes and the “junk DNA.” These are called CpG islands and are believed to help regulation of gene activity.
Unlike the human’s seemingly random distribution of gene-rich areas, genomes of many other organisms are more uniform, with genes evenly spaced throughout. Humans have on an average three times as many kinds of proteins as the fly or worm because of “alternative splicing” of messenger RNA (mRNA). This process can yield different protein products from the same gene that transcribes mRNA. Humans share most of the same protein families with worms, flies, and plants, but the number of gene family members has expanded in humans, especially in case of proteins involved in development and immunity. The human genome has a much greater portion (50%) of repeat sequences compared to other organisms for e.g., mustard weed (11%), the worm (7%), and the fruit fly (3%).
Scientists have proposed many theories to explain evolutionary contrasts between humans and other organisms, including those of life span, litter sizes, inbreeding, and genetic drift.
3.3 BENEFITS OR APPLICATIONS OF HUMAN GENOME PROJECT
Benefits derived from human genome sequencing are enormous and a few of the applications are mentioned below. There are exceptional opportunities to develop genomic research commercially with production and sale of DNA-based products and technologies that are useful for the following fields.
1 Molecular Medicine : New era of molecular medicine and biotechnology have emerged from the knowledge derived from HGP. Molecular medicine instead of treating a disorder based on the symptoms, it digs at the root causes of diseases. It aims at developing rapid and more accurate diagnostic tests or genetic screening for early detection of many diseases that enables effective treatment especially for single gene disorders. In addition it looks into genetic factors causing susceptibilities to common complex conditions (like diabetes, hypertension, heart disease, etc.,) in conjecture to environmental conditions and habits/addiction of the persons to smoking etc. Such information will help in assessing the extent of risk and predict the likely onset of a disorder even before it is expressed in an individual. That is it enables “Preclinical” or “Pre-symptomatic” diagnosis by using DNA probes (short stretches of DNA sequences synthesized with base sequence that is complementary to the target gene sequence) that are specifically designed for the detection of different diseases/disorders even before they are expressed. It is also possible to replace the defective genes by the normal genes by the method called “Gene Therapy”. In this method the normal gene or a target DNA sequence is incorporated into a vector (bacterial plasmid/a virus/liposome etc.,) and then transferred to patient’s tissue grown in culture. Once the target sequence is transfected i.e. incorporated into the recipient cells they are tested for expression of the transferred sequence or gene and then the tissue is grafted back into the patient where the incorporated normal gene will start functioning and the disease symptoms would disappear. Further, using genome sequence data novel therapeutic regimen can be developed using new classes of drugs, immunotherapy techniques and supplementing with the missing or defective protein.
2 Risk Assessment : It is understood from human genome analysis that nucleotide differences exist between different individuals which may be associated with their susceptibility or resistance to disease causing factors. Such an information will also be useful in assessing health damage and risks caused by exposure of individuals to radiations including long term low dose exposures and exposure to chemicals and toxins that induce harmful mutations and cancers and infections. This knowledge will help in modulating necessary preventive measures to maintain general health status and healthy society.
3 Energy and Environment : DOE initiated in 1994 for the Microbial genome Programme to sequence the genomes of bacteria which provide knowledge to benefit human health and environment apart from improving economy from industrial applications. Characterisation of complete microbial genomes will lead to the development of new energy related biotechnologies a) like photosynthetic systems, b) production of biofuels, c) microbial systems that work in extreme environments and also d) organisms that can metabolise readily available renewable resources and waste material. It is possible to develop diverse new products, processes and test methods that would help in maintaining pollution free environment. Above all knowledge of bacterial genomes helps pharmaceutical industries to identify how the pathogenic microbes cause diseases, in detecting new drug targets, identify the minimum number of genes necessary for maintaining life process and stand as models for understanding biological interactions and evolutionary history.
4 Anthropology, Evolution and Human Migration : Genomic information facilitated a) understanding of human evolution through germ line mutations in lineages b) knowing common biology the humans share with all other life c) study migration pattern of different population groups based on female genetic inheritance d) trace lineage and migration of males through the study of Y chromosome e) identify mutations and compare breakpoints in the evolution of mutations with ages of populations and historical events.
5 Forensic Science: Genome sequences are species specific and unique to different individuals. Hence genome sequence information used in forensic science is used for a) identifying victims who committed crimes, b) exonerate persons who are wrongly accused, c) identify crime and catastrophic victims d) establish paternity and identify relatives in cases of disputed parentage and e) matching the organ donors with that of recipient for organ transplantation. In addition identification of endangered and protected species among the wild life can be identified by analyzing their genomes of such species. It is possible to detect bacteria and other organisms that may pollute air, water, soil and food. The genomic information also helps in determining pedigrees of plant and livestock in breeding experiments.
6 Agriculture and Livestock Breeding Drought : Understanding of plants and human genomes allows the creation of disease resistant plants and more nutritious and pesticide free foods. Already the bioengineered seeds that are insect, pests and drought resistant are being marketed. Similarly disease resistant live stocks and those that are more productive for meat and milk yield are also being developed using genome information.
3.4 DISADVANTAGES OF HGP
The HGP which yielded enormos benefits for scientific research and mankind also led to fears and concerns about the information generated specially about an individual affected with genetic disease for which diagnostic or predictive tests are availble. The major disadvantage is the discrimination by the fellowmen and society which an individual suffers when affected with a genetic disorder. Such individuals are deprived of insurance coverage and will have to face difficulties to meet the medical bills which could be exorbitant. Further they may lose employment opportunities and those employed may be fired by the employers as they fear that an affceted employee may create safety risk at the work place, to the customers and also other employees specially when the genetic condition affects the coordination and judgement as in case of some neurological disorders. While the genetic screening can benefit a family by providing measures for preventing the recurrence among other members, it can also destroy the marriages and family relationships. There are also chances of misusing the genomic information by persons with selfish motives and destructive attitude. This will have a tremendous negative effect. In addition to the government, researchers and scientists, people from all walks of life should realise the negative effects and curb them as HGP offers heaps of benefits to the mankind and we have to reap the benefits it offers.
NEED FOR INDIVIDUAL DIPLOID HUMAN GENOME SEQUENCE
Originally HGP aimed at developing haploid reference genome that comprises 3.2 billion nucleotides. Other groups like International HapMap project, Appied Biosystem, Illumina, J. Craig Venter Institute(JCVI), Personal genome Project and Roche undertook the extension of obtaining reference sequence of diploid human genomes. On September 4th, 2007 Craig Venter’s complete DNA sequence was published unveiling for the first time the 6 billion nucleotide genome (diploid) of a single individual. His genome was sequenced from the 32 million sequence reads or more than 20 billion base pairs of DNA produced. The diploid genome sequences uniquely catalogued the contributions of the parental chromosomes (in which two sets of chromosomes one from his father and the other from his mother are represented) showing the amount of variation existing between the two. The human reference genome (HuRef) analysis now revealed that:
- The human to human variation is 5-7 times greater as compared to that reported in the earlier haploid genome analysis. This works out to a difference of 15-30 million base pairs between individuals.
- There are 4.1 million DNA variants in an individual of which 22% are nonSNP variants (RFLPs, VNTRs and microsatellites) but they account for about 74% of all the variants found in the DNA.
- There are 3.2 million SNPs and nearly non-SNP variants that include indels (insertion/deletion of nucleotides), copy number variants, block substitutions and segmental duplications. In Venter’s genome there were 1.2 million variants that were never before reported.
Analysis of diploid genome generated more informed haplotype assemblies. Haplotypes are linked variations found along the chromosomes (i.e. a set of alleles of different genes located on the same homologue with defined distance). The average occurrence of several haplotypes is reported in populations but not in individuals. Information on individual haplotypes enables study of rare or “private” variants which helps in predicting the traits and diseases in that person. This allows personalised medicine for treating a disease in an individual.
ETHICAL, LEGAL AND SOCIAL IMPLICATIONS (ELSI)
The ethical and social issues associated with the Genome Project have spurred a great deal of concern from the general public and researchers working within the project itself. These concerns lead to the formation of the ELSI committee of the human genome project. In 1997, the ELSI project received 11 million dollars of the Human genome budget, to study the implications of the Human Genome Project.(5) The ELSI committee has looked at many interrelated issues associated with the genome project. One such issue that has been investigated is the potential for gene therapy. Gene therapy involves locating a defective allele in an individual and replacing it with the correct allele. The genomic library created by the genome project will provide a way to determine what the correct sequence is at a particular loci and the correct base pairs may be able to be inserted into the affected individual if technology advances to that point. This has caused concern with many people because they feel that in a way mankind will be altering nature by eliminating the defective alleles from a population. Some say that by doing that we are in a sense creating super humans that have few if any genetic defects. This raises ethical concerns because some religious groups feel that mankind would be taking the role of the supreme creator if gene therapy becomes a reality. Currently molecular genetics has been able to identify metabolism defects due to a genetic alterations, the metabolite is then synthesized in a lab and injected into the affected individual thus relieving them of the disease condition. This too may have ethical implications because we are correcting a deficiency that was present at birth, thus altering the course of nature.
Discrimination based on genotype has also been raised. For example, employers may obtain genetic information on employees prior to hiring them. If a certain employee is shown to be genetically susceptible to undesirable work force traits they may be discriminated against. The same case is also possible with insurance companies. Health insurance companies may gain access to genetic information on an individual and thus discriminate against them, by refusing coverage or charging higher insurance premiums. If an individual is a carrier of alleles which have been associated with a particular disease it may make obtaining health insurance nearly impossible for the afflicted individual. If a genetic linkage is discovered that links alcohol abuse with a certain allele, auto insurance companies may also discriminate against such individuals by charging a higher premium. Congress has already enacted legislation banning discrimination based on genetic knowledge, so this may not be a major issue regarding the Human Genome Project.(6)
The Genome project may answer questions regarding evolution. By comparing human DNA with primate DNA, it may be possible to piece together the evolution puzzle. The possibility of unraveling the evolution puzzle has caused great controversy from religious groups around the world. If it is proven scientifically that evolution did in fact occur in humans, it would shake the ground work from which many religions are based on. This would have serious implications on the beliefs that have held true for centuries. Many feel that this is an area that should be left alone and not explored, due to the sensitivity of the potential findings. Although, if it were to be found that evolution did take place from the primates, it could possibly expand the potential uses of primates as genetic test subjects. If there was similar DNA at a specific loci, genetic tests could be performed to determine gene action and this could be applicable to human molecular genetics.
The genome project will also lead to improved techniques of genetic screening for diseases prior to birth. The genomic library created by the Genome Project will contain genetic information of fairly homogenous regions of the human genome, therefore there won’t be a great deal of variability between individuals at these loci. If a genetic disease is found to be produced from a specific mutation or allele being present, screening can occur to determine who these individuals are. By analyzing the gamete DNA from parents, it is possible to determine the probability a child might inherit genetic disorders from the parents. This can be adventitious from the standpoint of potential gene therapies. It could allow genetic disorders to be corrected before birth. This has sparked the debate over whether or not it is ethical to pre-select the genotype of the child they have. If genetics advances to the point of rapid selection and insertion of DNA into human individuals, “designer” children could potential become a reality. Parents may want to select children that were genetically predisposed for intelligence, athleticism or some other desirable trait.
Genetic tests may also be beneficial to people that have mutations at certain loci that make them particularly susceptible to an environmentally induced disease. Thus, they could potentially avoid the disease by avoiding the environment that could cause the disorder.
Another concern with the Human Genome Project relates to the monetary returns generated by it. It has been asked many times, “Who will receive the monetary benefits of this research if anyone?” This has been a major debate ever since the conception of the Human Genome Project, because the majority of the research is being funded by federal funds. The American society seems to feel that since they have funded the research through their tax dollars, they should receive this information free of charge. There have been numerous private companies that have begun sequencing the genome in hopes that they will be able to receive a great deal of financial gain from it. Thus the question remains, “Who owns the information generated by the project?” Already a trend has begun where a private company will patent a gene that it sequences. Many feel that such important information regarding the essence of life should be available to all mankind without a cost, but unfortunately the research required to obtain that information is very costly. Regardless of the issue involved, it has become apparent that intense regulation will have to be put in place, to insure that the information gained from the Human Genome Project is used correctly and for the benefit of all mankind. Such regulatory agencies should be comprised of individuals representing different facets of the debate. This way all viewpoints can be expressed, and all concerns can be heard.
Further Information
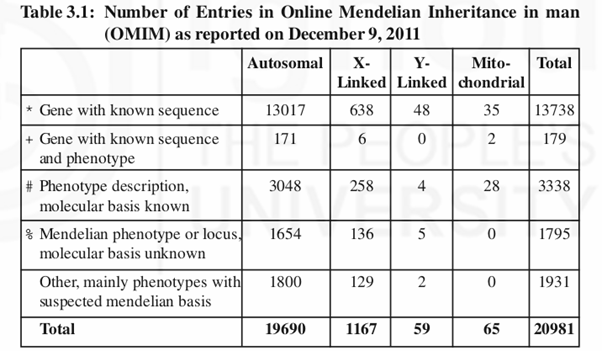
The life processes in any living organism are controlled by a set of genes that are located on chromosomes which are present in numbers that are highly specific for a given species. In humans there are 23 chromosomes present as pairs in all somatic cells (referred as diploid number or 2n) and as a single unit in gametic cells (referred as haploid number or n). Of the 23 pairs of chromosomes present in an individual, one set is inherited from the father and another from the mother along with the genes carried by them. Hence we see the resemblance of characters between the parents and their children. In human system there are trillion cells of different types that are organised into various tissues/organs that carry out myriad functions related to day to day life processes. All the functions carried out by these cells are controlled by genes located on the 23 chromosomes (table-3.1a and 3.2b).
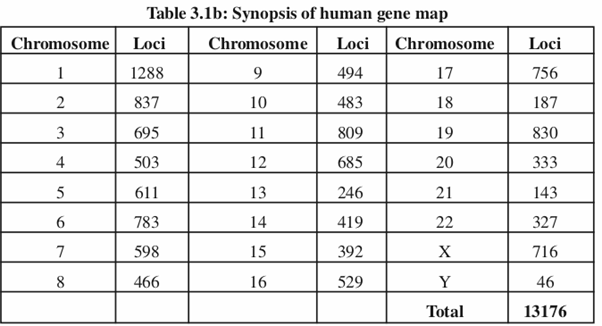

Physical location of all the genes on dif ferent chromosomes of an organism is represented as “Genetic Maps”. These maps are generated by determining distances between different genes present on a chromosome by an approach called linkage analysis [Box-3.1]. The distance between two genes is expressed as CentiMorgan (cM) which is a unit of genetic distance. Thus 1cM represents 1% probability of recombination occurring during meiosis i.e. gametogenesis. The genes present on the same chromosome are said to be linked and the group of such linked genes are called linkage groups or chromosomes.
Chemically genes are made up of a macromolecule called Deoxyribose Nucleic Acid (DNA) which exists as a double helical structure resembling a ladder. Chemically, DNA comprises 4 nitrogenous bases- Adenine (A), Guanine (G), Thymine (T) and Cytosine (C) which are arranged as rungs of the ladder and supported by a sugar-phosphate backbone (Fig.3.1). Each base with a sugar and phosphate molecule is referred as a nucleotide. The nitrogenous bases Adenine and Guanine are referred as “Purines” and Thymine and Cytosine as “Pyrimidines”. This structure of DNA as described by Watson and Crick (1953) satisfies all the criteria of a genetic material including the segregation of different genes/characters through generations
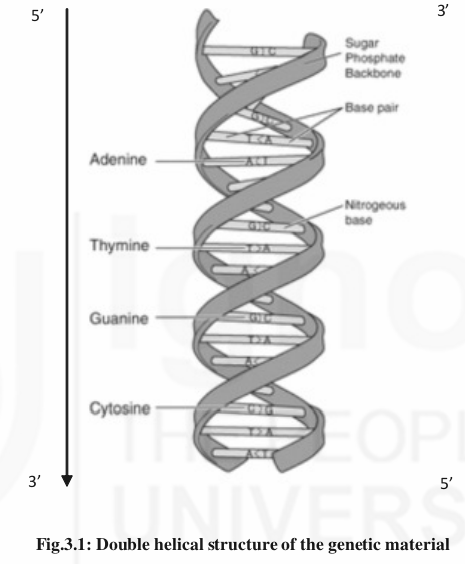
The structure of de-oxyribose nucleic acid (DNA) comprising 4 nitrogenous bases (Adenine, Guanine, Thymine, Cytosine) each attached to a sugar and phosphate molecule that form a backbone. Adenine and Guanine are called purines and Thymine and Cytosine as pyrimidines. The base pairing is strictly complementary i.e. Adenine always pairs with Thymine while Guanine always pairs with Cytosine. The bases are held together by hydrogen bonds forming rungs of the ladder. The two strands of the DNA forming twisted double helical structure run in the opposite directions i.e. one strand runs from 5’ to 3’ and the other from 3’ to 5’direction.
In early 1950s human geneticists have attempted to map some disease genes using certain genetic markers like ABO blood groups. One such study established close linkage between the loci for a disease called Nail-Patella syndrome and that of ABO blood groups. In the following years researchers attempted to map several other disease genes using different polymorphic loci related to serum proteins, enzymes and leucocyte (HLA) antigens. Later with the discovery of enzymes called restriction endonucleases or restriction enzymes (REs), new markers known as restriction fragment length polymorphisms (RFLPs) were identified which proved to be better markers in genetic analysis experiments.
Restriction enzymes cut the DNA at specific sites breaking them into fragments of different sizes. If a given DNA sequence has one restriction site 2 fragments of different lengths will be generated. The number of fragments generated will be n+1 when n number of restriction enzymes is used to cut the given DNA sequence. In later years certain sequences of nucleotides or base pairs (bps) were found to be repeated in various numbers differing in different individuals thus showing polymorphism. These stretches of repeats (0.1-20kb long) are referred to as variable number of tandem repeats (VNTRs) or minisatellites where the core repeat sequence of DNA carries 15 to hundreds of nucleotides. Initially DNA finger printing – a technique followed in forensic science used certain VNTR markers. Later smaller stretches of repeat sequences (<0.1 kb) called microsatellites with only 1-4 nucleotides (occurring as di, tri and tetranucleotide repeats) in each stretch were identified which are highly polymorphic. Now more than 6000 such markers located on different chromosomes are available for conducting any study. VNTRs and microstaellite markers were extensively used in 1990s in gene mapping studies and studies on their associations with diseases and risk predictions. With the discovery of single nucleotide polymorphisms (SNPs read as snips) which distinguishes individuals at single nucleotide level, research in human genetics took a different turn with the application of genome wide screening for mapping genes, finding differences between population groups, between normal and disease samples which in turn aids in formulating better treatment measures.
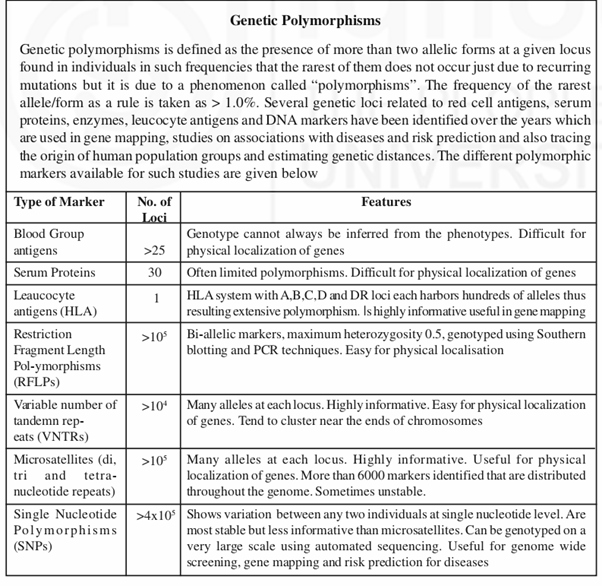
In spite of the availability of thousands of polymorphic markers (RFLPs, VNTRs, Microsatellites and snips), generation of human genetic map by mapping all the estimated 30,000 genes one by one appeared to be a Herculean task in that it is both tedious and time consuming. Hence to overcome this difficulty, the idea of determining the entire sequence of nucleotides in the DNA was floated which was discussed in depth at several scientific meetings before it was finally approved and the stage to undertake the Human Genome Project (HGP) was set.